Sprawdź się
The EU Joint Programme – Neurodegenerative Disease Research (JPND) jest największym na świecie programem naukowym zajmującym się walką z chorobami neurodegeneracyjnymi. W obszarze badań JPND znajdują się:
choroba Alzheimera;
choroba Parkinsona;
choroby prionowe;
stwardnienie zanikowe boczne;
choroba Huntingtona;
ataksja rdzeniowo‑móżdżkowa;
rdzeniowy zanik mięśni.
Jedną z powyższych chorób opisano po raz pierwszy w 1817 r. Początkowo nadano jej nazwę drżączki poraźnej. Objawami są m.in. spowolnienie ruchowe (bradykinezja), sztywność mięśni i drżenie kończyn (drżenie spoczynkowe). Nie jest to choroba genetyczna. Bezpośrednią przyczyną objawów jest uszkodzenie neuronów istoty czarnej mózgowia.
Indeks górny Źródło: informacje zamieszczone na portalu Neurodegeneration Research. Indeks górny koniecŹródło: informacje zamieszczone na portalu Neurodegeneration Research.
- 65–75 lat; Wartość: 1000000; Udział procentowy: 17,2%
- 75–84 lata; Wartość: 2700000; Udział procentowy: 46,6%
- >85 lat; Wartość: 2100000; Udział procentowy: 36,2%
Indeks górny Źródło: Alzheimer’s Association, 2020 Alzheimer’s Disease Facts and Figures. Alzheimer’s Association Report, „Alzheimer’s & Dementia. The Journal of the Alzheimer’s Association” 2020, nr 16(3), s. 391–460. Indeks górny koniecŹródło: Alzheimer’s Association, 2020 Alzheimer’s Disease Facts and Figures. Alzheimer’s Association Report, „Alzheimer’s & Dementia. The Journal of the Alzheimer’s Association” 2020, nr 16(3), s. 391–460.
Wybierz spośród poniższych wszystkie prawidłowy wniosek wyciągnięty z danych zobrazowanych na wykresie. Możliwe odpowiedzi: 1. Osoby powyżej 85 roku życia są mniej narażone na zachorowanie niż osoby w przedziale wiekowym 75 – 84 lata., 2. Wśród osób przechodzących z przedziału wiekowego 75 – 84 lata do 85+ obserwuje się spadek zachorowalności na chorobę Alzheimera., 3. Wśród osób chorych w USA, najmniejszy odsetek stanowią osoby poniżej 75 roku życia., 4. Osoby cierpiące na chorobę Alzheimera powyżej 85 roku życia stanowią większy odsetek populacji niż zdrowe osoby powyżej 85 roku życia.
Chorobami autoimmunologicznymi nazywamy schorzenia, w których przebiegu limfocyty układu immunologicznego pacjenta niszczą komórki i tkanki własnego organizmu. Przykładami takich chorób są miastenia oraz ostre rozsiane zapalenie mózgu i rdzenia. W przypadku pierwszej z tych chorób limfocyty atakują receptory acetylocholinowe znajdujące się na płytkach motorycznych (nerwowo‑mięśniowych) chorego. Uszkodzenie tych receptorów powoduje, że acetylocholina wydzielona przez zakończenie neuronu ruchowego nie wywołuje odpowiedniej reakcji (skurczu) mięśnia poprzecznie prążkowanego. Objawami choroby są m.in. opadanie powiek i nieprawidłowe działanie mięśni mimicznych.
W przypadku ostrego rozsianego zapalenia mózgu i rdzenia dochodzi do ataku limfocytów na glikolipid o nazwie mielina, wchodzący w skład otoczki mielinowej. Objawy tej choroby są zbliżone do obserwowanych w przebiegu stwardnienia rozsianego, jednak występują głównie u dzieci.
Indeks górny Na podstawie: Sagmin Lee i in., A Potential Link Between Autoimmunity and Neurodegeneration in Immune‑Mediated Neurological Disease, „Journal of Neuroimmunology” 2011, nr 235(1–2), s. 56–69. Indeks górny koniecNa podstawie: Sagmin Lee i in., A Potential Link Between Autoimmunity and Neurodegeneration in Immune‑Mediated Neurological Disease, „Journal of Neuroimmunology” 2011, nr 235(1–2), s. 56–69.
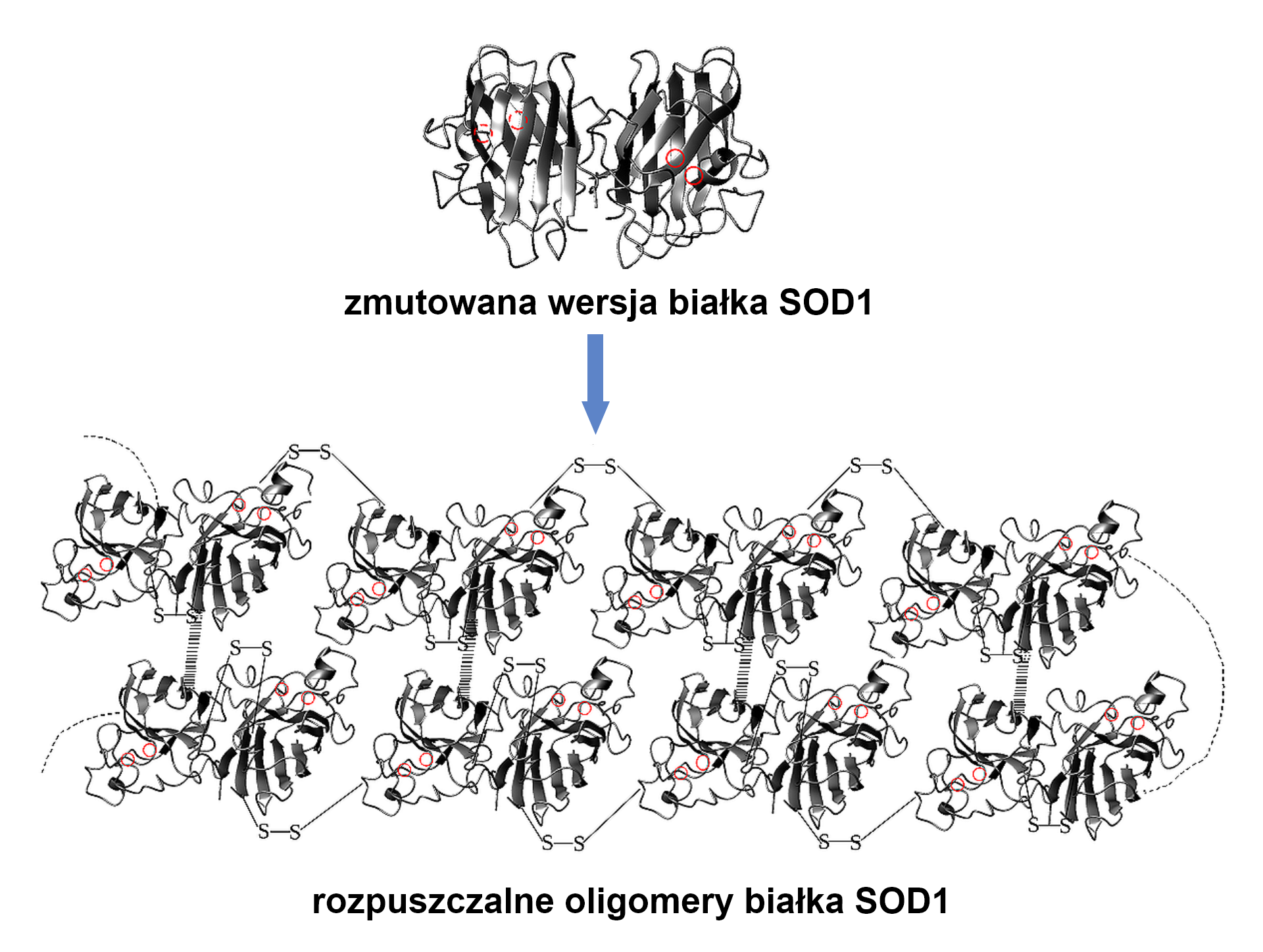
W przebiegu wielu chorób neurodegeneracyjnych w tkance nerwowej dochodzi do gromadzenia się szkodliwego, nierozpuszczalnego białka zwanego amyloidem (nazwa pochodzi od struktury przypominającej amylazy i amylopektyny skrobi). Stwardnienie zanikowe boczne (SLA – sclerosis lateralis amyotrophica) powodowane jest przez toksyczne oligomery białka SOD1, powstające w sposób podobny do amyloidów. Między pojedynczymi jednostkami białka SOD1 tworzą się wiązania skutkujące powstaniem rozpuszczalnych oligomerów, transportowanych do mitochondrium. Tendencja do takiego łączenia się białek SOD1 jest konsekwencją licznych mutacji w genie kodującym to białko. Mutacje powodujące powstanie nieprawidłowego białka SOD1 wywołują taką zmianę jego struktury, że niemożliwe jest utrzymanie jonów cynku i miedzi (obecnych w białku prawidłowym).
Indeks górny Źródło: Lucia Banci i in., SOD1 and Amyotrophic Lateral Sclerosis: Mutations and Oligomerization, „PLOS ONE” 2008, nr 3(2), s. e1677. Indeks górny koniecŹródło: Lucia Banci i in., SOD1 and Amyotrophic Lateral Sclerosis: Mutations and Oligomerization, „PLOS ONE” 2008, nr 3(2), s. e1677.
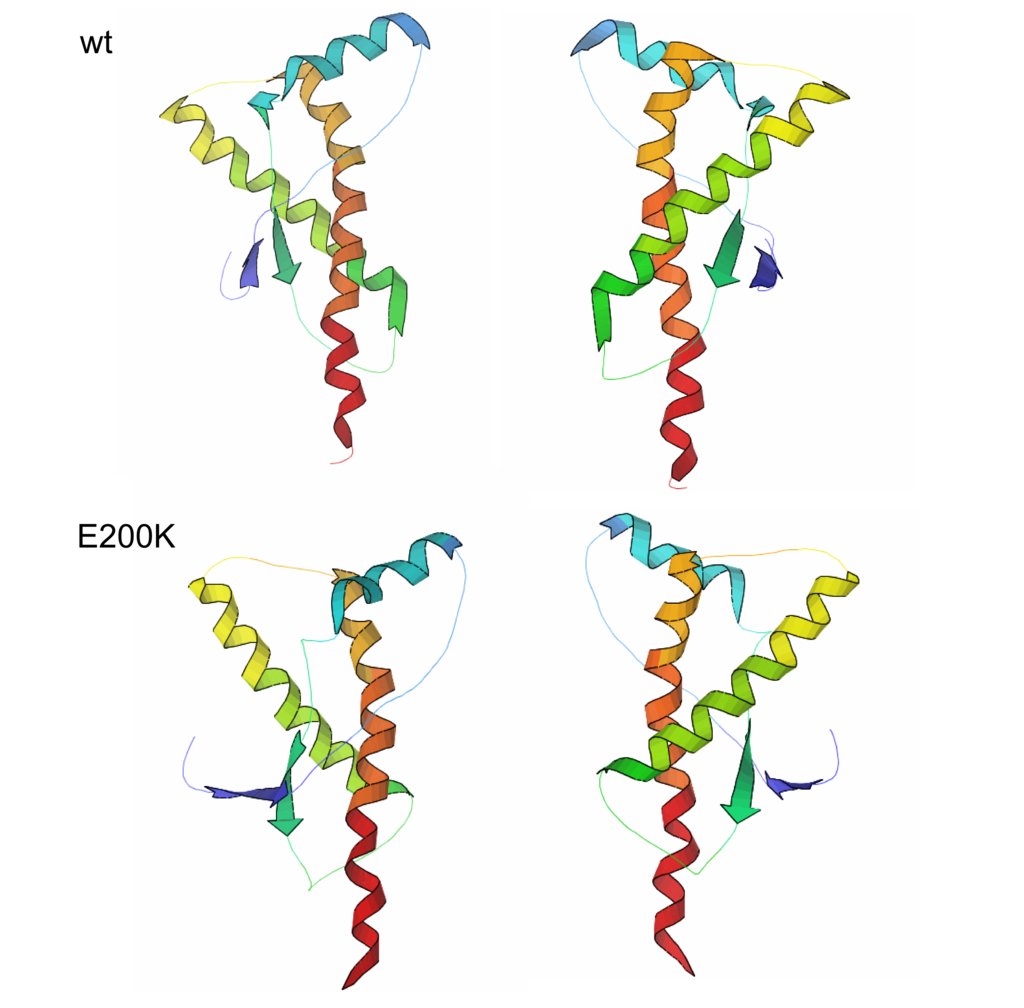
Priony to uszkodzone wersje prawidłowych białek obecnych w neuronach. Często mają identyczną z białkami prawidłowymi budowę pierwszorzędową (sekwencję aminokwasów), ale inną budowę drugo- i trzeciorzędową (występują różnice w strukturze trójwymiarowej białek). Choroba Creutzfeldta–Jakoba (z ang. CJD – Creutzfeldt‑Jakob disease) jest neurodegeneracyjną chorobą prionową. Występuje w kilku wersjach:
samoistnej – źródło prionów pojawiających się w mózgu nie jest znane;
jatrogennej – priony pochodzą z zanieczyszczonych preparatów hormonalnych pochodzących z ludzkich przysadek mózgowych;
wariancie CJD (vCJD) – do zakażenia dochodzi najprawdopodobniej po spożyciu podrobów wołowych wyprodukowanych z bydła zakażonego encefalopatią gąbczastą bydła (BSE); objawy choroby rozwijają się jedynie u osób mających specyficzny genotyp;
rodzinnej – spowodowanej odziedziczeniem od rodzica mutacji w genie kodującym białko PrP.
Białka prionowe, w sposób nie do końca poznany, zmieniają strukturę drugo- i trzeciorzędową prawidłowych białek mózgu PrP, przekształcając je w priony (PrP SC). Skutkuje to uszkodzeniem neuronów.
Indeks dolny Źródło: Creutzfeldt‑Jakob Disease, Classic (CJD), portal Centers for Disease Control and Prevention. Indeks dolny koniecŹródło: Creutzfeldt‑Jakob Disease, Classic (CJD), portal Centers for Disease Control and Prevention.
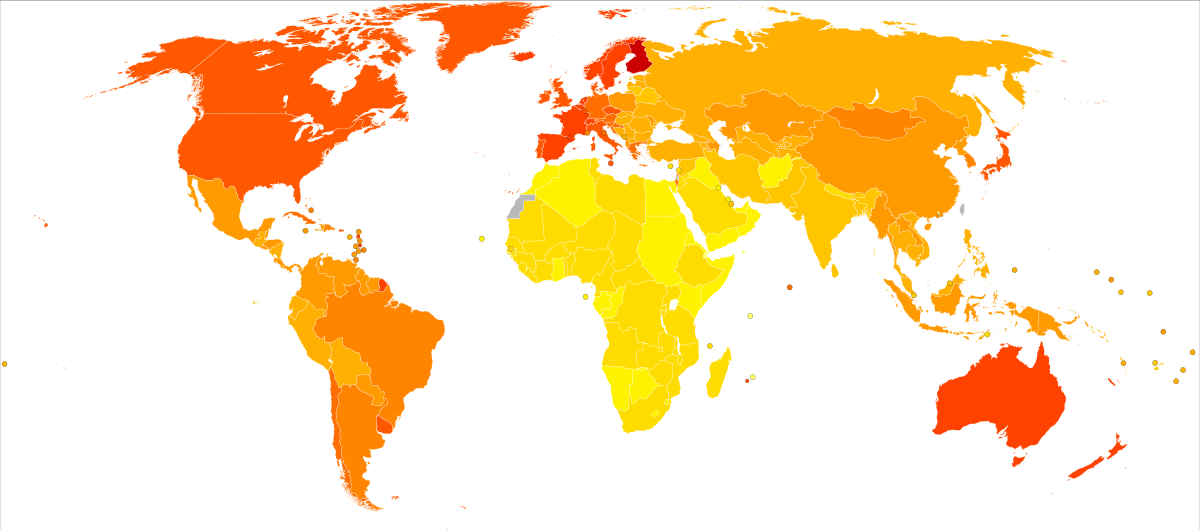
Powyższa mapa jest graficzną prezentacją liczby osób ze zdiagnozowaną chorobą Alzheimera i innymi rodzajami demencji na 100 tys. obywateli. Im bardziej intensywny kolor na mapie, tym więcej przypadków choroby: kolor intensywnie czerwony oznacza stan powyżej 300 chorych na 100 tys. mieszkańców, kolor jasnożółty – poniżej 100 chorych na 100 tys. mieszkańców.