Biotechnologia i jej zastosowania
Inżynieria genetyczna - narzędzia i techniki
Przedstawia narzędzia wykorzystywane w biotechnologii molekularnej: enzymy: polimerazy, ligazy i enzymy restrykcyjne oraz określisz ich zastosowania.
Przedstawisz istotę technik stosowanych w inżynierii genetycznej: hybrydyzacji DNA, analizy restrykcyjnej i elektroforezy DNA, metody PCR, sekwencjonowania DNA metodą Sangera.
Rozwój biologii molekularnej w drugiej połowie XX wieku zapoczątkował gwałtowny postęp w badaniach nad funkcjonowaniem materiału genetycznego. Jednym z najważniejszych osiągnięć tego okresu było opracowanie technik inżynierii genetycznej, które umożliwiły bezpośrednią ingerencję w DNA organizmów. Dzięki nim naukowcy mogą izolować geny, modyfikować ich sekwencje, przenosić je między organizmami oraz kontrolować ich ekspresję
Enzymy wykorzystywane w biotechnologii molekularnej
Wprowadzenie zmian w DNA wymaga wykorzystania enzymów. Do najważniejszych z nich należą:
polimerazy DNA,
enzymy restrykcyjne (restryktazy),
ligazy.
Enzymy charakteryzują się specyficznością substratową. Oznacza to, że mają zdolność do łączenia się z konkretnymi substratami i katalizują reakcje po odpowiednim dopasowaniu substratu do centrum aktywnego enzymu. Efektem tego zjawiska jest precyzyjna kontrola procesów zachodzących w komórkach.
Ligazy są grupą enzymów, które katalizują powstawanie nowych wiązań pomiędzy nukleotydami w niciach DNA z wykorzystaniem energii w postaci ATP. Lgazy łączą pęknięte nici DNA, np. przecięte enzymami restrykcyjnymi za pomocą wiązań fosfodiestrowych. Proces ten, nazywany ligacją, jest wydajniejszy, jeżeli dotyczy cząsteczek DNA z lepkimi końcami.

Tworzenie hybrydowego DNA
Dzięki restryktazom i ligazom można precyzyjnie wyciąć wybrany gen z jednego organizmu i wstawić go do innej cząsteczki DNA, zwanej wektorem. Wektor to nośnik DNA – najczęściej mała, kolista cząsteczka DNA bakterii zwana plazmidem, która potrafi się samodzielnie namnażać w komórce. Wycięty fragment DNA, który chcemy wprowadzić do wektora, nazywamy wstawką.
Aby połączyć wstawkę z plazmidem, oba fragmenty tnie się tym samym enzymem restrykcyjnym. Powstają wtedy tzw. „lepkie końce”, czyli pasujące do siebie fragmenty DNA, które mogą się połączyć. Następnie ligazaskleja je, tworząc jedną cząsteczkę. DNA zawierający fragmenty pochodzące od co najmniej dwóch różnych organizmów (np. plazmid bakteryjny i wstawka z DNA wirusowego) nazywany jest DNA hybrydowym (zrekombinowanym).
Przed wykonaniem takiego eksperymentu przeprowadza się analizę restrykcyjną. Polega ona na sprawdzeniu, w których miejscach dana restryktaza przetnie DNA. Tworzy się tzw. mapę restrykcyjną, czyli schemat pokazujący miejsca cięcia w plazmidzie. Dzięki temu wiadomo, którego enzymu użyć i gdzie dokładnie DNA zostanie przecięte. Pozwala to zaplanować wstawienie genu w odpowiednie miejsce.

Wydajność ligacji jest o wiele większa w przypadku lepkich końców, ponieważ mogą one łączyć się wiązaniami wodorowymi z komplementarnymi jednoniciowymi końcami innych cząsteczek DNA.
Analiza restrykcyjna
Aby sprawdzić, czy doszło do połączenia (ligacji) wstawki z plazmidem oraz czy gen został wstawiony w odpowiedniej orientacji, wykonuje się analizę restrykcyjną. Polega ona na przecięciu uzyskanego hybrydowego DNA tymi samymi enzymami, których użyto do przygotowania wstawki i wektora.
W dalszym etapie, powstałe po restrykcji fragmenty należy rozdzielić i porównać ich wielkość. Służy do tego elektroforeza DNA, która pozwala precyzyjnie odróżnić plazmid z nowym genem od plazmidu „pustego” na podstawie ich różnej długości.
Elektroforeza DNA
Elektroforeza to metoda polegająca na poruszaniu się naładowanych cząstek w polu elektrycznym. Cząstki o ładunku dodatnim dążą do elektrody ujemnej (katody), mające zaś ładunek ujemny – do elektrody dodatniej (anody).
Cząsteczki kwasów nukleinowych (DNA i RNA) posiadają grupy fosforanowe, przez co mają ładunek ujemny. W efekcie migrują one w stronę bieguna dodatniego, a szybkość tego procesu zależy od ich wielkości – im krótsza cząsteczka DNA lub RNA, tym szybciej przemieszcza się ona w kierunku anody.
Elektroforeza żelowa pozwala na rozdzielenie kwasów nukleinowych w porowatym żelu. Żel to substancja o konsystencji galarety, składająca się z agarozy lub poliakrylamidu. Wylewa się go do specjalnej formy, w której tworzy cienką warstwę. Struktura żelu działa jak sito, umożliwiając separację cząsteczek w polu elektrycznym.
W żelu znajdują się studzienki, w których umieszcza się próbki (np. plazmidy po trawieniu enzymami restrykcyjnymi). Oprócz badanych cząsteczek, na żel nanosi się również marker masy (tzw. drabinkę DNA – ang. DNA ladder), czyli mieszaninę fragmentów kwasu nukleinowego o znanej długości, która służy jako układ odniesienia.
Żel umieszcza się w aparacie do elektroforezy wypełnionym buforem, a następnie poddaje działaniu pola elektrycznego. Pod jego wpływem fragmenty kwasów nukleinowych migrują od katody w stronę anody z różną szybkością. Mniejsze cząsteczki przemieszczają się szybciej, gdyż łatwiej przeciskają się przez pory żelu, podczas gdy cząsteczki znacznie większe od porów pozostają niemal nieruchome.
Ponieważ DNA jest bezbarwne, do próbek dodaje się barwnik, który pozwala śledzić postęp elektroforezy. Aby jednak uwidocznić same prążki DNA, stosuje się barwniki fluorescencyjne (np. bromek etydyny), które wymagają obserwacji w świetle UV. Proces zazwyczaj prowadzi się pod napięciem 100–120 V przez około 20–30 minut.
Po zakończonej elektroforezie żel analizuje się w świetle UV. Rozdzielone fragmenty widoczne są w postaci prążków dzięki świeceniu barwników fluorescencyjnych wiążących się z DNA.
Wielkość fragmentów DNA określa się na podstawie porównania ich szybkości migracji w żelu (na podstawie pozycji odpowiednich prążków) z szybkością migracji fragmentów DNA o znanej długości (pozycje prążków z drabinki).
Liczba prążków świadczy o tym, ile miejsc restrykcyjnych zostało rozpoznanych przez enzymy restrykcyjne w danej cząsteczce DNA.
Przeprowadź doświadczenie w wirtualnym laboratorium „Analiza restrykcyjna i obserwacja produktów rozdziału elektroforetycznego uzyskanych fragmentów restrykcyjnych”. Następnie odpowiedz na polecenie.
Przeprowadź doświadczenie w wirtualnym laboratorium biotechnologicznym. Rozwiąż problem badawczy i zweryfikuj hipotezę. W formularzu zapisz swoje obserwacje, sformułuj wnioski i zweryfikuj hipotezy.
W celu produkcji białka X stworzono wstawkę z genem tego białka zakończoną lepkimi końcami. Przeprowadzono ligację wstawki z plazmidem (wektorem) w miejscu rozpoznawanym przez enzym HindIII. Ligacja może dawać trzy różne rezultaty:
brak wbudowania wstawki,
wstawka wbudowana w prawidłowej orientacji,
wstawka wbudowana w odwrotnej orientacji (nie ulega ekspresji).
Plazmidy namnożono w koloniach bakteryjnych, a następnie wyizolowano, otrzymując 5 próbek zawierających wyizolowane plazmidy (po jednej próbce z każdej kolonii bakteryjnej). W celu przeprowadzania analizy restrykcyjnej próbki trawiono enzymem PvuI. Na podstawie długości powstałych fragmentów można wnioskować o wyniku ligacji.
Problem badawczy: Które plazmidy po ligacji zawierają wstawki w odpowiedniej orientacji?
Hipoteza 1: Wszystkie plazmidy zawierają prawidłowo wbudowane wstawki.
Hipoteza 2: Wstawka została prawidłowo wbudowana tylko w niektóre plazmidy.
Materiał biologiczny:
plazmidy zawierające geny X uzyskane z kolonii bakteryjnych
Sprzęt laboratoryjny:
probówki Eppendorfa;
pipeta automatyczna z końcówkami;
aparat do elektroforezy;
kuweta z lodem.
Odczynniki:
woda destylowana;
bufor do trawienia z barwnikiem;
enzym restrykcyjny PuvI;
marker masy;
żel do elektroforezy.
Zasób interaktywny dostępny pod adresem https://zpe.gov.pl/a/DEL5DQA25
Laboratorium 1
Przeprowadzono doświadczenie w wirtualnym laboratorium biotechnologicznym.
W celu produkcji białka X stworzono wstawkę z genem tego białka zakończoną lepkimi końcami i przeprowadzono ligację wstawki z plazmidem (wektorem) w miejscu rozpoznawanym przez enzym HindIII. Ligacja może dawać trzy różne rezultaty - są to brak wbudowania wstawki, wstawka wbudowana w prawidłowej orientacji (pod promotorem) oraz wstawka wbudowana w odwrotnej orientacji (nie ulega ekspresji). Plazmidy namnożono w koloniach bakteryjnych. W celu przeprowadzania analizy restrykcyjnej próbki trawiono enzymem PvuI. Na podstawie długości powstałych fragmentów można wnioskować o wyniku ligacji.
Problem badawczy: Czy plazmidy po ligacji zawierają wstawki w odpowiedniej orientacji?
Hipoteza 1: Wszystkie plazmidy zawierają prawidłowo wbudowane wstawki.
Hipoteza 2: Nie do wszystkich plazmidów udało się wbudować wstawkę w dobrej orientacji.
Materiał biologiczny: kolonie bakteryjne zawierające plazmidy po ligacji genu X.
Sprzęt laboratoryjny: probówki Eppendorfa, pipeta automatyczna z końcówkami, aparat do elektroforezy, kuweta z lodem.
Instrukcja wykonania doświadczenia:
Szczegóły doświadczenia 1
Bufor do trawienia zawiera jony magnezu, które są kofaktorami enzymów transkrypcyjnych.
Szczegóły doświadczenia 2
Bufor do trawienia zawiera również barwnik, dzięki któremu próbki rozdzielone na żelu są widoczne w świetle UV.
Szczegóły doświadczenia 3
W elektroforezie rozdział cząsteczek zachodzi zależnie od ich wielkości: mniejsze cząsteczki migrują szybciej niż duże. Marker masy zawiera fragmenty DNA o znanych długościach, dzięki czemu porównując układ prążków z próbek oraz z markera można oszacować długość fragmentów obecnych w próbkach.
Obserwacje: Odczytaj długość uzyskanych fragmentów w każdej próbce, porównując ich położenie z markerem masy.
Wnioski: Porównaj długość uzyskanych fragmentów z długością wzorcowych fragmentów zamieszczonych na schemacie powyżej.
Obserwacje: W kolejnych próbkach długość fragmentów uzyskanych w wyniku analizy restrykcyjnej wynosi kolejno:
1. 900 pz, 1000 pz, 2000 pz;
2. 500 pz, 900 pz i 2500 pz;
3. 900 pz, 1800 pz; 4. 500 pz, 900 pz i 2500 pz;
5. 500 pz, 900 pz i 2500 pz.
Wnioski: Wstawka wbudowała się prawidłowo w próbkach 2, 4 i 5, a w odwrotnej orientacji w próbce 1. W próbce 3 ligacja nie zaszła.
Weryfikacja hipotez: Hipoteza 1 jest nieprawdziwa, a hipoteza 2 jest prawdziwa.
Zastosowanie elektroforezy
Elektroforezę DNA stosuje się m.in. w biologii molekularnej, farmakologii, medycynie sądowej, weterynarii, diagnostyce medycznej oraz kontroli jakości żywności. Wykorzystywana może być także w kryminalistyce do identyfikowania osób podejrzanych o popełnienie przestępstwa.
Sondy molekularne i hybrydyzacja kwasów nukleinowych
Sonda molekularna to krótki, jednoniciowy fragment kwasu nukleinowego (DNA lub RNA) o znanej sekwencji nukleotydów, który służy do wykrywania określonych, komplementarnych do niego sekwencji w badanej próbie materiału genetycznego.
Ważną cechą sond molekularnych jest ich znakowanie, czyli przyłączanie określonych substancji (tzw. znaczników), które umożliwiają ich detekcję oraz wykrycie komplementarnych do nich fragmentów kwasów nukleinowych. W tym celu wykorzystuje się fluorofory – substancje o zdolności do fluorescencji w świetle UV. Wśród nich wyróżnić można takie związki jak fluoresceinafluoresceina, rodaminarodamina czy cyjaninycyjaniny.

Sondy molekularne działają w oparciu o zjawisko hybrydyzacji, czyli parowania się ze sobą komplementarnych nici kwasów nukleinowych. Są wykorzystywane do wykrywania:
konkretnych genów w genomie - hybrydyzacja Southern blot,
produktów ekspresji konkretnych genówu - hybrydyzacja northern blot,
sekwencji DNA bezpośrednio w chromosomach lub jądrach interfazowych - fluorescencyjna hybrydyzacja in situ - FISH.
Znakowaną sondę o znanej sekwencji dodaje się do mieszaniny jednoniciowych cząsteczek DNA lub RNA, a następnie poddaje inkubacji. Jeśli w badanej próbce znajduje się poszukiwana sekwencja, sonda trwale się z nią łączy (hybrydyzuje). Ponieważ sonda posiada znacznik (np. cząsteczkę fluorescencyjną), wynik sprawdza się poprzez detekcję sygnału – tam, gdzie pojawi się sygnał znacznika, tam znajduje się szukany gen lub jego produkt.
Typ hybrydyzacji | Cel | Materiał | Typ analizy | Typ sondy |
Southern blot | określenie lokalizacji lub wykrywanie obecności danej sekwencji DNA | wyizolowane DNA | DNA‑DNA | sondy DNA |
|---|---|---|---|---|
Northern blot | identyfikacja określonej sekwencji RNA | wyizolowane RNA | RNA‑DNA | sondy DNA lub RNA |
Fluorescencyjna hybrydyzacja in situ (FISH) | wykrywanie określonej sekwencji DNA | nienaruszone chromosomy, komórki | DNA‑DNA | sondy DNA |
Przeanalizuj animację „Sondy molekularne i hybrydyzacja DNA”, a następnie wykonaj polecenia.

Film dostępny pod adresem /preview/resource/R1KtqOLtmTtH9
Film opisuje sondy molekularne i zjawisko hybrydyzacji DNA.

Modyfikacją klasycznej hybrydyzacji jest metoda mikromacierzy, polegająca na rozmieszczeniu sond molekularnych na podłożu zwanym mikromacierzą (ang. microarray). Może to być płytka szklana lub plastikowa, na której nanoszone są w regularnych odstępach sondy, różniące się sekwencjami. Do tych sond mogą przyłączać się cząsteczki DNA lub RNA, które będą wykazywały się wysokim stopniem komplementarności. W przeciwieństwie do innych metod, w przypadku mikromacierzy znakowany jest materiał badany, a nie sondy.
Dzięki miniaturyzacji technika wykorzystania mikromacierzy ta pozwala na jednoczesną analizę ekspresji wielu, a czasami wszystkich genów w dwóch probówkach (np. pacjenta zdrowego i chorego). Materiał znajdujący się w obu próbkach znakowany jest różnymi barwnikami, tak aby możliwe było ich odróżnienie. Takie próbki kwasu nukleinowego hybrydyzuje się z mikromacierzą. Cząsteczki wyznakowanego kwasu nukleinowego wiążą się do komplementarnych sekwencji sond na mikromacierzy.
Odczyt mikromacierzy wykonywany jest za pomocą metody obrazowania umożliwiającej ilościowy pomiar sygnału fluorescencyjnego. Intensywność sygnału dla poszczególnych sond mikromacierzy jest proporcjonalna do ilości w próbce kwasu nukleinowego o danej sekwencji związanego z sondą. W przypadku braku różnic w ekspresji genów widoczna barwa będzie wypadkową obu znaczników. Przewaga jednego z kolorów świadczy o większej ekspresji w probówce zawierającej barwnik o danej fluorescencji.
Zastosowanie hybrydyzacji
Technika hybrydyzacji wykorzystywana jest w diagnostyce nowotworów oraz chorób zakaźnych i o podłożu genetycznym (m.in. fenyloketonurii, hemofilii). W badaniach naukowych metody oparte na hybrydyzacji stosuje się w celu analizy struktury kwasów nukleinowych, identyfikacji sekwencji powtórzonych oraz śledzenia aktywności komórek. Hybrydyzacja DNA pozwala również na określenie pokrewieństwa ewolucyjnego, a także identyfikację żywności genetycznie zmodyfikowanej. Co więcej, metoda ta jest wykorzystywana do monitorowania zmian środowiska, w tym określania bioróżnorodności mikroorganizmów.
Reakcja łańcuchowa polimerazy (PCR)
Reakcja łańcuchowa polimerazy - PCR (ang. Polymerase Chain Reaction) to technika powielania fragmentu DNA w ilościach sięgających milionów, a nawet miliardów kopii w krótkim czasie.
Metoda ta została opracowana w USA w 1983 r. przez zespół Kary’ego Mullisa, za którą Mullis w 1993 r. otrzymał Nagrodę Nobla.
Reakcję PCR przeprowadza się w termocyklerachtermocyklerach mieszaninie reakcyjnej o zdefiniowanym składzie. Polega ona na wielokrotnym powtarzaniu określonych etapów, które zachodzą w różnych temperaturach, a każda zmiana temperatury wyznacza koniec danego etapu.
W skład mieszaniny reakcyjnej wchodzi matrycowe DNA, które służy jako podstawa do namnożenia określonego fragmentu. Może to być DNA genomowe wyizolowane z danego organizmu, plazmid, izolat DNA z próbki biologicznej o nieznanej sekwencji czy wcześniej uzyskany fragment DNA po działaniu restryktaz.
W zależności od celu wykorzystania reakcji PCR projektuje się odpowiednie startery (primery), które pozwalają na wyznaczenie obu końców powielanej sekwencji DNA. Startery to krótkie sekwencje DNA, zawierające zazwyczaj ok. 20 nukleotydów.
Startery umożliwiają przyłączenie polimerazy DNA– enzymu syntetyzującego nową nić na matrycy DNA. W reakcji PCR używa się termostabilnych polimeraz, takich jak polimeraza Taq (z bakterii Thermus aquaticus), która wykazuje maksymalną aktywność w temp. 75–80°C. Jest to szczególnie ważna cecha – denaturację DNA w reakcji PCR osiąga się przez ogrzanie mieszaniny do temperatury 95–98°C, a więc takiej, w której inne polimerazy ulegają inaktywacji. Podczas syntezy nowych nici DNA polimeraza zużywa nukleotydy, które dostarczone są w postaci mieszaniny. Mieszanina ta często opisywana jest jako dNTP (trifosforany deoksyrybonukleozydowetrifosforany deoksyrybonukleozydowe), a w jej skład wchodzą: dATP, gGTP, dCTP oraz dTTP, odpowiadając kolejno: nukleotydowi adeninowemu, guaninowemu, cytozynowemu i tyminowemu.
Przebieg PCR
Reakcja PCR składa się z trzech etapów:
denaturacji,
przyłączania,
elongacji.
Wszystkie trzy etapy powtarza się cyklicznie (zazwyczaj od 25 do 35 razy). Ponieważ każda nowa nić staje się matrycą w kolejnym cyklu, liczba kopii DNA rośnie wykładniczo. W efekcie w ciągu krótkiego czasu można otrzymać miliony identycznych fragmentów DNA, których długość i końce są precyzyjnie wyznaczone przez sekwencje użytych starterów.
Zalety i wady PCR
Zastosowanie reakcji PCR
Możliwość szybkiego powielenia materiału genetycznego w reakcji PCR umożliwia jej wykorzystanie w wielu dziedzinach. Jest jednym z podstawowych narzędzi w biotechnologii; służy m.in. do namnażania (amplifikacji) genów, genotypowaniagenotypowania, wykrywania mutacji czy identyfikacji mikroorganizmów.
W medycynie reakcja PCR znalazła zastosowanie w diagnostyce chorób (poprzez wykrywanie genów organizmów patogennych i związanych z nowotworzeniem)czy ustalaniu rodzicielstwa.
Technika PCR to także przydatne narzędzie w walce z przestępczością. Analiza śladów DNA pozostawionych na miejscu przestępstw pozwala na wykluczenie lub wskazanie potencjalnego sprawcy, a także ustalenie tożsamości osób zaginionych lub odnalezionych zwłok.
Modyfikacja reakcji PCR, określana jako RT‑qPCRRT‑qPCR (ilościowa reakcja łańcuchowej polimerazy z odwrotną transkrypcją) pozwala na analizę ekspresji genów.
Utrwal informacje o reakcji PCR wykonując doświadczenie w wirtualnym laboratorium:
Przeprowadź doświadczenie w laboratorium. Rozwiąż problem badawczy i zweryfikuj hipotezę. Zapisz uzyskane wyniki w formularzu, a następnie ustal wnioski.
Zapoznaj się z opisem doświadczenia w laboratorium, a następnie zweryfikuj hipotezy.
Temat: Amplifikacja fragmentu DNA metodą PCR
Problem badawczy: Czy badana próbka zawiera DNA o szukanym fragmencie?
Hipoteza 1: W badanej próbie jest obecne DNA o danej sekwencji.
Hipoteza 2: W badanej próbie nie występuje DNA o tej sekwencji.
Sprzęt laboratoryjny:
probówki Eppendorfa
termocykler
pipeta automatyczna
kuweta z lodem
aparat do elektroforezy
Materiały
bufor 10x stężony (odpowiedni dla danej polimerazy) zawierający 15 mM MgClIndeks dolny 22
nukleotydy dNTP (dATP, dCTP, dTTP, dGTP; mieszanina nukleotydów o stężeniu 10 mM każdy)
startery: 10 M każdy
matryca (np. DNA plazmidowy, całkowity genomowy DNA)
woda destylowana sterylna
polimeraza Taq
Ilustracja przedstawia laboratorium. Znajdują się w nim: aparat do elektroforezy, kuweta z lodem - w kuwecie znajdują się probówki Eppendorfa, termocykler, pipeta automatyczna, zlewka z wodą destylowaną, cylinder miarowy, kolby. W laboratorium do tablicy korkowej jest przypięta kartka papieru zawierająca dwie tabele z ilościami składników mieszaniny. Tabela pierwsza jest z trzema kolumnami: skład mieszaniny, próba badana podana w mikrolitrach, kontrola negatywna podana w mikrolitrach. Skład mieszaniny, woda: próba badania 40 mikrolitrów, kontrola negatywna 41 mikrolitrów. Skład mieszaniny, bufor: próba badania 5 mikrolitrów, kontrola negatywna 5 mikrolitrów. Skład mieszaniny, dNTP (10 nanometrów każdy): próba badania 1 mikrolitr, kontrola negatywna 1 mikrolitr. Skład mieszaniny, starter 1 (10 mikrometrów): próba badania 1 mikrolitr, kontrola negatywna 1 mikrolitr. Skład mieszaniny, starter 2 (10 mikrometrów): próba badania 1 mikrolitr, kontrola negatywna 1 mikrolitr. Skład mieszaniny, DNA (100 nanogramów na mikrolitr): próba badania 1 mikrolitr, kontrola negatywna brak danych. Skład mieszaniny, DNA (100 nanogramów na mikrolitr): próba badania 1 mikrolitr, kontrola negatywna brak danych. Skład mieszaniny, polimeraza DNA: próba badania 1 mikrolitr, kontrola negatywna 1 mikrolitr. Tabela druga jest z trzema kolumnami. Wskazana jest w niej: 1. Denaturacja wstępna przy dziewięćdziesięciu czterech stopniach Celsjusza, trwająca 5 minut. 2. Denaturacja przy dziewięćdziesięciu czterech stopniach Celsjusza, trwająca 10 sekund i 30 cykli. 3. Przyłączenie przy pięćdziesięciu pięciu stopniach Celsjusza, trwające 30 sekund i 30 cykli. 4. Wydłużanie przy siedemdziesięciu dwóch stopniach Celsjusza, trwające jedną minutę i 30 cykli. 5. Końcowe wydłużanie przy siedemdziesięciu dwóch stopniach Celsjusza, trwające pięć minut. Instrukcja wykonania doświadczenia: 1. Za pomocą pipety automatycznej umieszczono odpowiednie ilości składników mieszaniny reakcyjnej, zgodnie z protokołem podanym na kartce. 2. Umieszczono próbę badaną i kontrolę negatywną w termocyklerze. 3. Uzupełniono program reakcji PCR. 4. Za pomocą pipety przeniesiono próbę badaną i kontrolę negatywną do aparatu do elektroforezy i uruchomiono go. 5. Porównano wynik elektroforezy próby badanej i kontrolnej. Wyniki są następujące: Na żelu po elektroforezie w próbie badanej widoczny jest wyraźny prążek, który nie występuje w kontroli negatywnej. W próbie badanej jest obecne DNA o szukanej sekwencji, które uległo powieleniu w wyniku reakcji PCR. W kontroli negatywnej reakcja nie zaszła, bo nie dodano DNA matrycowego.
Sekwencjonowanie DNA metodą Sangera
Sekwencjonowanie DNA jest jedną z podstawowych metod genetyki molekularnej, polegającą na odczytywaniu kolejności nukleotydów w nici DNA.
Sekwencjonowanie DNA przeprowadza się kilkoma metodami. Jedną z nich jest metoda enzymatyczna, znana również jako metoda Sangera (od nazwiska jej twórcy, Fredericka Sangera) lub metodą terminacji łańcucha. Pomimo upływu lat, po licznych modyfikacjach, technika ta jest z powodzeniem stosowana do dziś.
Cechą charakterystyczną metody Sangera jest wykorzystanie dideoksynukleotydów (ddNTP), które pełnią rolę „stoperów” podczas tworzenia nowej cząsteczki DNA. Są to tzw. nukleotydy terminalne. W normalnym DNA nukleotydy (dNTP) mają grupę -OH, która pozwala na dołączenie do nich kolejnego nukleotydu. Dideoksynukleotydy (ddNTP) nie posiadają tej grupy. W rezultacie, gdy polimeraza DNA wbuduje ddNTP zamiast zwykłego nukleotydu, proces wydłużania nici zostaje przerwany. W efekcie powstaje zestaw fragmentów DNA o wszystkich możliwych długościach, z których każdy kończy się specyficznym ddNTP.
Sekwencjonowanie metodą Sangera prowadzi się w czterech probówkach o różnym składzie:
probówka „A”: 4 nukleotydy → A, C, T, G, ddATP, polimeraza DNA;
probówka „G”: 4 nukleotydy → A, C, T, G, ddGTP, polimeraza DNA;
probówka „C”: 4 nukleotydy → A, C, T, G, ddCTP, polimeraza DNA;
probówka „T”: 4 nukleotydy → A, C, T, G, ddTTP, polimeraza DNA..
Po zakończeniu sekwencjonowania przeprowadzana jest elektroforeza, która ma na celu pokazanie nukleotydów terminalnych, czyli znajdujących się na końcu każdej z nici DNA. Po naświetleniu żelu, na którym są rozdzielone nukleotydy każdej z czterech probówek, można uzyskać obraz wyłącznie nukleotydów dołączonych na nowo i pokazać je na chromatogramie.

Mitochondrialny genom człowieka zsekwencjonowano już w 1981 r., co przyczyniło się do podjęcia próby poznania sekwencji całego genomu człowieka. Wyniki Projektu poznania ludzkiego genomu (ang. Human Genome Project), trwającego od 1990 do 2003 r., opublikowano w 2001 r. Poznanie sekwencji genomu nie stanowiło jednak zakończenia całego procesu. Następnym zadaniem było udzielenie odpowiedzi na pytanie o rolę danych sekwencji, a także ich wpływ na funkcjonowanie genomu. Badania te prowadzone są nadal.
Utrwal wiadomości o sekwencjonowaniu przeprowadzając sekwencjonowanie w wirtualnym laboratorium.
Przeprowadź doświadczenie w pracowni genetycznej. Rozwiąż problem badawczy, zapisz wyniki, sformułuj wnioski i zweryfikuj hipotezę.
Prawidłowa sekwencja początkowego fragmentu genu łańcucha beta hemoglobiny:
GTG CAT CTG ACT CCT GAG GAG
Temat: Sekwencjonowanie genu łańcucha beta hemoglobiny metodą Sangera
Problem badawczy: Czy sekwencja genu łańcucha beta hemoglobiny w pobranej próbce DNA jest prawidłowa?
Hipoteza 1: Sekwencja genu łańcucha beta hemoglobiny w badanej próbce jest prawidłowa.
Hipoteza 2: Sekwencja genu łańcucha beta hemoglobiny w badanej próbce nie jest prawidłowa.
Sprzęt laboratoryjny:
4 puste probówki Eppendorfa na statywie
pipeta automatyczna o objętości 10–100 mul
żel agarozowy
Odczynniki:
próbka z matrycowym DNA
mieszanina dideoksynukleotydów
ddATP znakowany żółtym barwnikiem
ddCTP znakowany niebieskim barwnikiem
ddGTP znakowany czerwonym barwnikiem
ddTTP znakowany zielonym barwnikiem
polimeraza DNA
Zasób interaktywny dostępny pod adresem https://zpe.gov.pl/a/DEL5DQA25
Laboratorium 1
Przeprowadzono doświadczenie w pracowni genetycznej.
Prawidłowa sekwencja początkowego fragmentu genu łańcucha beta hemoglobiny: GTG CAT CTG ACT CCT GAG GAG.
Temat: Sekwencjonowanie genu łańcucha beta hemoglobiny metodą Sangera
Problem badawczy: Czy sekwencja genu łańcucha beta hemoglobiny w pobranej próbce DNA jest prawidłowa?
Hipoteza 1: Sekwencja genu łańcucha beta hemoglobiny w badanej próbce jest prawidłowa.
Hipoteza 2: Sekwencja genu łańcucha beta hemoglobiny w badanej próbce nie jest prawidłowa.
Sprzęt laboratoryjny: cztery puste probówki Eppendorfa na statywie; pipeta automatyczna o objętości od 10 do 100 mul; żel agarozowy.
Odczynniki: próbka z matrycowym DNA; mieszanina dideoksynukleotydów; ddATP znakowany żółtym barwnikiem; ddCTP znakowany niebieskim barwnikiem; ddGTP znakowany czerwonym barwnikiem; ddTTP znakowany zielonym barwnikiem; polimeraza DNA.
Instrukcja:
1. Przygotowano mieszaninę reakcyjną do reakcji łańcuchowej polimerazy PCR. Do czterech probówek Eppendorfa dodano za pomocą pipety automatycznej:
- 5 mikrolitrów matrycy DNA - z radioaktywnie wyznakowanym starterem;
- 10 mikrolitrów mieszaniny deoksynukleotydów;
- 3 mikrolitry dideoksynukleotydów (do każdej z probówek Eppendorfa inny rodzaj: ddATP, ddCTP, ddGTP, DDTTP).
2. Włożono cztery probówki Eppendorfa zawierające mieszaniny reakcyjne do termocyklera i przeprowadzono PCR.
3. Po zakończeniu PCR pobrano pipetą automatyczną po 10 mikrolitrów próbek Eppendorfa. Umieszczono je w studzienkach w żelu agarozowym. Przeprowadzono elektroforezę DNA.
4. Odczytano sekwencję DNA bezpośrednio z żelu. Wykonano ćwiczenia interaktywne.
Szczegóły zjawiska 1
Przyłączenie dideoksynukleotydu uniemożliwia dalsze wydłużanie nici DNA przez polimerazę, ponieważ jest on pozbawiony grupy –OH w pozycji 3′, więc nie ma możliwości utworzenia wiązania fosfodiestrowego z kolejnym nukleotydem.
Szczegóły zjawiska 2
Do sekwencjonowania stosuje się wysoko rozdzielczą elektroforezę żelową, która umożliwia rozróżnienie nawet tych łańcuchów DNA, które różnią się tylko jednym nukleotydem.
Szczegóły zjawiska 3
Stężenie deoksynukleotydu musi być znacznie wyższe niż stężenie odpowiedniego dideoksynukleotydu, aby umożliwić wytworzenie wystarczającej liczby fragmentów przy jednoczesnej transkrypcji całej sekwencji.
Wyniki: DNA pochodzący z badanej próbki ma sekwencję: GTG CAT CTG ACT CCT GTG GAG.
Wnioski: Fragment DNA zawiera mutację punktową w szóstym kodonie genu podjednostki beta hemoglobiny.
Weryfikacja hipotezy: Hipoteza 1 jest fałszywa. Hipoteza 2 jest prawdziwa.
Zastosowanie sekwencjonowania DNA
Sekwencjonowanie DNA jest często wykorzystywane w medycynie, a także biotechnologii i biologii molekularnej. Poznanie sekwencji DNA wybranego organizmu i zawartych w tym fragmencie informacji pozwala na ulepszenie i przyspieszenie diagnostyki chorób, a także bardziej spersonalizowaną terapię. Sekwencjonowanie DNA w medycynie znajduje zastosowanie także przy identyfikacji podłoża chorób nowotworowych.
Wektory i klonowanie DNA
Klonowanie DNA to proces polegający na namnożeniu (powieleniu) danego fragmentu DNA w komórkach, zwykle bakterii Escherichia coli, przy zastosowaniu specjalnego nośnika DNA – wektora genetycznego.
Wektory genetyczne
Wektor połączony z fragmentem DNA, który ma zostać skopiowany (najczęściej zawierający konkretny gen lub geny) tworzy cząsteczkę zrekombinowanego DNA.
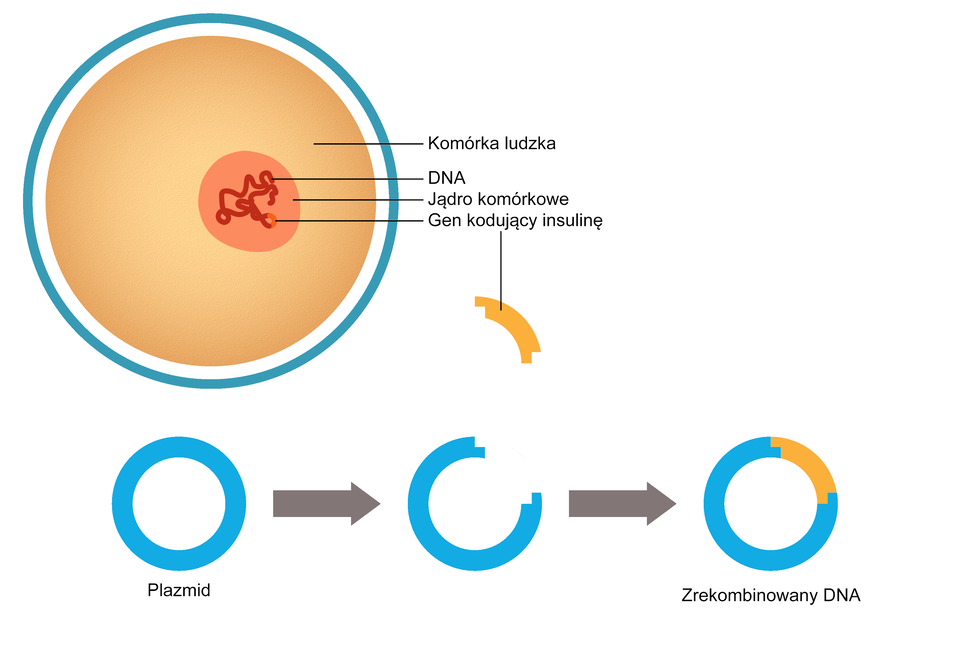
Jako wektory genetyczne wykorzystuje się plazmidy, sztuczne chromosomy oraz różne typy wirusów, w tym fagi dla bakterii oraz wektory wirusowe dla komórek eukariotycznych.
Aby cząsteczka DNA mogła być wektorem genetycznym musi posiadać następujące cechy:
zawierać miejsce rozpoznawane przez enzymy restrykcyjne;
zawierać marker selekcyjnymarker selekcyjny, pozwalający na selekcję komórek gospodarza zawierających wektor;
zawierać miejsce inicjacji replikacji tzw. ori;
musi być łatwa do izolowania z komórek gospodarza.
Wektory genetyczne, ze względów etycznych, nie powinny mieć genów, które mogą stanowić zagrożenie ekologiczne.
Zastosowanie wektorów genetycznych
Wektory genetyczne są wykorzystywane przede wszystkim do klonowania DNA.
Zapoznaj się z filmem samouczkiem „Wektory genetyczne”, a następnie wykonaj polecenia.

Film dostępny pod adresem /preview/resource/R1Dp0J5TGDX4B
Nagranie filmowe pod tytułem Wektory genetyczne.
Klonowanie DNA
Do przeprowadzenia klonowania DNA niezbędne są:
docelowy fragment DNA zawierający gen, który ma zostać powielony (sklonowany) do komórki;
wektor genetyczny – służący jako nośnik fragmentu DNA przeznaczonego do sklonowania;
enzymy restrykcyjne – służące do wycięcia wybranego fragmentu DNA z genomu organizmu, z którego zostanie pobrany, oraz przecięcia wektora, np. plazmidu; enzymy te rozpoznają swoiste sekwencje w dwuniciowej cząsteczce DNA, nazywane miejscami restrykcyjnymi, i przecinają DNA w danym miejscu;
ligaza – enzym umożliwiający połączenie wyciętego fragmentu DNA z DNA wektora;
gen selekcyjny – znacznik pokazujący, czy procedura się powiodła.
Etapy klonowania DNA
Klonowanie DNA można podzielić na dwa etapy:
otrzymanie zrekombinowanego DNA poprzez wyizolowanie określonego genu i przeniesienie go do wektora;
wprowadzenie wektora ze wstawionym genem do wybranej komórki i powielenie go na skutek replikacji.
Po zakończeniu klonowania DNA przeprowadza się selekcję, czyli wyodrębnienie bakterii, które przyjęły zrekombinowany plazmid wraz z genem reporterowym zapewniającym oporność na antybiotyk, od tych, które go nie mają. Po przeprowadzonej transformacji bakterie posiewa się na płytki z podłożem selekcyjnym z antybiotykiem. Bakterie bez plazmidu zginą; te zaś, które zyskały dzięki nowym genom antybiotykooporność, będą rosnąć na pożywkach z dodatkiem odpowiedniego antybiotyku, tworząc małe kolonie. Każda z nich będzie zawierać taki sam zrekombinowany plazmid z klonowanym fragmentem DNA.
Aby się upewnić, że uzyskane kolonie mają plazmid o prawidłowej strukturze, sprawdzane są pod kątem obecności zrekombinowanego genu. W tym celu wykorzystuje się reakcję PCR przeprowadzaną na wyizolowanych plazmidach z kilku kolonii lub trawienie za pomocą odpowiednich enzymów restrykcyjnych. O obecności klonowanego DNA świadczy uzyskanie fragmentów DNA o określonej długości.
Zastosowanie klonowania DNA
Techniki klonowania molekularnego są powszechnie stosowane w badaniach DNA, genów, ich funkcji oraz ekspresji. Dzięki nim możliwe stały się identyfikacja oraz izolacja genów, które są odpowiedzialne za występowanie różnych chorób. Stanowi to podłoże do zastosowania terapii genowej. Co więcej, sama terapia genowa wykorzystuje techniki klonowania DNA poprzez dostarczenie funkcjonalnych fragmentów DNA do komórek pacjentów.
Klonowanie DNA pozwala także na uzyskanie organizmów transgenicznych do produkcji ludzkich białek, np. bakterii produkujących ludzkie hormony wzrostu czy insulinę. Wykorzystywane jest również w produkcji szczepionek, które uzyskuje się poprzez tworzenie odpowiednich białek.
Ponadto klonowanie umożliwia tworzenie bibliotek genomowych i bibliotek cDNA.
Zapoznaj się z animacją „Klonowanie DNA”. Wykonaj polecenie.

Film dostępny pod adresem /preview/resource/RsiuRGxNzfHGp
Film pod tytułem: Klonowanie DNA.
Biblioteki genomowe i biblioteki cDNA
W bibliotekach genowych, nazywanych też bankami genowymi, przechowywane są zestawy sklonowanych fragmentów DNA składających się na kompletny genom danego organizmu lub pełny zestaw produktów transkrypcji. Banki genowe dzieli się na:
biblioteki genomowe obejmujące zbiór fragmentów genomu organizmu powstały w wyniku klonowania DNA chromosomalnego i DNA organellarnego w wektorach genetycznych,
biblioteki cDNA zawierające fragmenty cDNA, czyli fragmenty DNA powstające po przepisaniu informacji ze wszystkich wyizolowanych cząsteczek mRNA (transkryptomy) na DNA przy pomocy enzymu o nazwie odwrotna transkryptaza.
Bank genów jest miejscem do poszukiwania i izolacji genu. Istnieją także komercyjne banki genowe specjalizujące się w przechowywaniu DNA ludzkiego.
Tworzenie bibliotek genomowych
Tworzenie biblioteki genomowej rozpoczyna izolacja DNA genomowego (gDNA) danego organizmu. Wyizolowane gDNA zostaje poddane trawieniu restrykcyjnemu za pomocą enzymów restrykcyjnych. Wolne końce uzyskanych w ten sposób fragmentów DNA łączone są z wektorem genetycznym ligazą DNA. Następnie stworzone wektory wprowadzane są do komórek bakteryjnych lub drożdżowych. Komórki te, dzieląc się, powielają wprowadzony do nich fragment DNA. Bardzo ważne jest, aby dany wektor zawierał wyłącznie jeden fragment DNA genomowego – jednak ze względu na losowość cięcia enzymami restrykcyjnymi fragmenty DNA w różnych komórkach mogą się na siebie nakładać.
Tworzenie bibliotek cDNA
Tworzenie biblioteki cDNAcDNA przebiega podobnie do tworzenia bibliotek genomowych, jednak wymaga innego przygotowania materiału do klonowania. W pierwszej kolejności niezbędna jest izolacja mRNA z komórek, tkanek lub całego organizmu. Ze względu na brak możliwości bezpośredniego połączenia wyizolowanego mRNA z DNA wektorów, niezbędne jest „przepisanie” mRNA na cDNA. Można tego dokonać dzięki enzymowi – odwrotnej transkryptazie – zdolnemu do syntezy nici DNA na bazie mRNA. Synteza drugiej nici zachodzi dzięki polimerazie DNA. Tak otrzymane dwuniciowe DNA poddaje się trawieniu restrykcyjnemu. Kolejne etapy przebiegają analogicznie jak przy tworzeniu bibliotek genomowych.

Biblioteki cDNA stanowią zgromadzone w wektorach i umieszczone w komórce bakterii cDNA, które ogranicza się wyłącznie do genów aktywnych transkrypcyjnie - nie zawierają intronów, dlatego tworzone wektory mogą być wykorzystywane w procesach klonowania genów eukariotycznych.
Zastosowanie bibliotek genowych
Podsumowanie
Inżynieria genetyczna rozwinęła się wraz z postępem biologii molekularnej i umożliwia bezpośrednią ingerencję w DNA, w tym izolowanie, modyfikowanie, klonowanie i analizę genów oraz kontrolę ich ekspresji.
Podstawowymi narzędziami biotechnologii molekularnej są enzymy:
- polimerazy DNA – katalizują syntezę DNA na matrycy DNA lub RNA (np. odwrotna transkryptaza); są kluczowe w reakcji PCR i analizie ekspresji genów;
- enzymy restrykcyjne – tną DNA w ściśle określonych sekwencjach, umożliwiając otrzymywanie fragmentów DNA, tworzenie rekombinowanego DNA oraz map restrykcyjnych;
- ligazy DNA – łączą fragmenty DNA poprzez tworzenie wiązań fosfodiestrowych, co pozwala na wstawianie genów do wektorów.Analiza restrykcyjna polega na trawieniu DNA enzymami restrykcyjnymi i identyfikacji powstałych fragmentów na podstawie ich długości; umożliwia sprawdzenie obecności i orientacji wstawki DNA w plazmidzie.
Elektroforeza DNA jest techniką rozdziału fragmentów kwasów nukleinowych w polu elektrycznym; pozwala porównywać długość fragmentów DNA na podstawie ich migracji w żelu i wizualizacji prążków.
Hybrydyzacja DNA wykorzystuje sondy molekularne do wykrywania komplementarnych sekwencji DNA lub RNA; obejmuje m.in. metody Southern blot, Northern blot, FISH oraz mikromacierze DNA.
Reakcja łańcuchowa polimerazy (PCR) umożliwia szybkie i wielokrotne powielanie wybranego fragmentu DNA; znajduje zastosowanie w diagnostyce medycznej, kryminalistyce, biologii molekularnej i biotechnologii.
Sekwencjonowanie DNA metodą Sangera pozwala na odczytanie kolejności nukleotydów w DNA; jest wykorzystywane w badaniach naukowych, diagnostyce chorób genetycznych i nowotworowych.
Klonowanie DNA polega na namnażaniu fragmentów DNA w komórkach gospodarza z użyciem wektorów genetycznych (np. plazmidów); prowadzi do otrzymania zrekombinowanego DNA.
Biblioteka genomowa - jest zbiorem sklonowanych fragmentów całego genomu organizmu; zawiera zarówno geny kodujące, jak i sekwencje niekodujące. Stosowana jest m.in. do badań struktury genomu i lokalizacji genów.
Biblioteka cDNA - jest zbiorem sklonowanych fragmentów DNA powstałych na podstawie mRNA wyizolowanego z określonej tkanki lub komórek; nie zawiera intronów ani sekwencji niekodujących, ponieważ cDNA powstaje z dojrzałego mRNA przy udziale odwrotnej transkryptazy. Jest wykorzystywana m.in. do analizy ekspresji genów oraz produkcji białek w organizmach prokariotycznych.
Biblioteki genomowe i biblioteki cDNA stanowią zbiory sklonowanych fragmentów DNA i są podstawowym źródłem do identyfikacji, izolacji i analizy genów, w tym genów aktywnych transkrypcyjnie.
Techniki inżynierii genetycznej mają kluczowe znaczenie w medycynie, biotechnologii, przemyśle i nauce, umożliwiając rozwój diagnostyki, terapii oraz badań nad funkcją genów
Ćwiczenia utrwalające

Wektory genetyczne stosuje się w celu... Możliwe odpowiedzi: 1. fragmentacji DNA., 2. przenoszenia i klonowania zrekombinowanego DNA., 3. otrzymywania organizmów transgenicznych., 4. pozyskiwania materiału genetycznego wykorzystywanego do tworzenia kolistych cząsteczek DNA.
Wróć do polecenia na stronie „Na dobry początek” i dopisz brakujące definicje. Pamiętaj, żeby nie kopiować słownika, ale wyjaśnić każde słowo kluczowe w miarę możliwości swoimi słowami.