Laboratorium kontroli jakości leków
MED.09. Sporządzanie i wytwarzanie produktów leczniczych oraz prowadzenie obrotu produktami leczniczymi, wyrobami medycznymi, suplementami diety i środkami spożywczymi specjalnego przeznaczenia żywieniowego oraz innymi produktami dopuszczonymi do obrotu w aptece na podstawie przepisów prawa - Technik farmaceutyczny 321301
Aparat do badania szybkości uwalniania substancji leczniczej z postaci leku
SYMULATOR
Badanie uwalniania substancji czynnej z postaci leku – definicja, zastosowanie
Uwalnianie substancji leczniczej (ang. active pharmaceutical ingredient – API) z postaci leku jest kluczowe dla jej biodostępności i skuteczności farmakologicznej. Proces ten zależy od wielu czynników, np. postaci leku, jakości, rodzaju API i jej właściwości fizykochemicznych (np. rozpuszczalności, stopnia rozdrobnienia, postaci krystalicznej), rodzaju i jakości substancji pomocniczych czy od przebiegu procesu wytwarzania. Z tego względu badanie uwalniania, określane również jako badanie dostępności farmaceutycznej, jest jednym z ważnych narzędzi do charakteryzowania preparatów farmaceutycznych. Pozwala ono ocenić w warunkach in vitro stopień i szybkość przejścia substancji leczniczej z postaci leku do otaczającego płynu i rozpuszczenia się w nim.
Badanie uwalniania może znaleźć zastosowanie m.in. w:
kontroli jakości:
na etapie rozwoju produktu (charakterystyka API i ocena nowych formulacji),
po rejestracji leku (rutynowe badanie udowadniające powtarzalność serii produkcyjnych na podstawie porównania wyznaczonej w badaniu szybkości uwalniania z wartością zamieszczoną w specyfikacji produktu, a także pozwalające ocenić wpływ wprowadzanych podczas produkcji zamierzonych, niewielkich zmian na właściwości produktu końcowego),
do oceny stabilności produktu (wykazanie, że podczas przechowywania leku nie zaszły żadne zmiany mogące mieć wpływ na biodostępność).
przewidywaniu biodostępności in vivo na podstawie badań in vitro (korelacja in vitro – in vivo), co jest bardzo pomocne w projektowaniu leku,
ocenie biorównoważności dwóch produktów, dzięki czemu można pominąć badania in vivo.
Aparatura
W celu właściwej oceny procesu uwalniania i uzyskania dokładnych i powtarzalnych wyników konieczna jest standaryzacja aparatów i ujednolicenie procedur wykonania badań dostępności farmaceutycznej. Opisy rodzajów aparatów, podstawowych parametrów i kryteriów interpretacji wyników badania uwalniania z różnych postaci leków są zawarte w farmakopeach. Wydanie XII Farmakopei Polskiej (FP XII) prezentuje cztery ustandaryzowane aparaty do badania uwalniania substancji leczniczej ze stałych doustnych postaci leków (o niemodyfikowanym, przedłużonym lub opóźnionym uwalnianiu, np. tabletek, kapsułek):
aparat 1 – koszyczkowy,
aparat 2 – łopatkowy,
aparat 3 – z ruchomym cylindrem,
aparat 4 – przepływowy.
Ponadto FP XII zawiera monografie przedstawiające opisy aparatów do badania procesu uwalniania z systemów transdermalnych, gum do żucia i lipofilowych stałych postaci leku.
Badanie uwalniania z kapsułek i tabletek najczęściej przeprowadza się z wykorzystaniem aparatów 1 i 2. Aparat koszyczkowy jest zalecany przy badaniu dostępności farmaceutycznej z postaci ulegających flotacji, np. kapsułek, tabletek flotacyjnych, a także postaci wymagających zmiany płynu akceptorowego w trakcie badania, np. tabletek o opóźnionym uwalnianiu. Dla form ulegających flotacji możliwe jest także wykorzystanie aparatu łopatkowego; w tym przypadku należy pamiętać jednak o dociążeniu każdej jednostki poprzez umieszczenie jej w obciążniku (tzw. sinkerach) lub dołączenie do niej np. drucianej spirali. Aparat łopatkowy może także zostać wykorzystany do badania np. zawiesin, proszków, a jeżeli jest wyposażony w dodatkowe specjalistyczne elementy (zgodne z monografią farmakopealną, takie jak dysk nośny, komorę ekstrakcyjną lub zamiast mieszadła łopatkowego w element mieszający z cylindrem), służy do badania systemów transdermalnych. Z kolei aparat przepływowy jest rekomendowany przede wszystkim dla form zawierających substancje trudno rozpuszczalne, ale także dla zawiesin. W przypadku aparatu przepływowego wyposażonego w specjalną komorę można badać uwalnianie API z lipofilowych stałych postaci leku (takich jak czopki, kapsułki miękkie), ponadto z mikrosfer i implantów. Aparat z ruchomym cylindrem wykorzystuje się m.in. do badania form nierozpadających się. Ostatnim typem aparatu do uwalniania opisanym w FP XII jest odpowiednio zaprojektowany aparat do przeprowadzenia badania leczniczych gum do żucia.
Dla niektórych postaci leków wykonanie badania dostępności farmaceutycznej jest obowiązkowe (jeżeli zostało to zapisane w odpowiednich monografiach), zaleca się jednak, aby przeprowadzać je dla wszystkich postaci leków wykazujących działanie ogólnoustrojowe.
Najpopularniejsze są aparaty łopatkowy i koszyczkowy, rzadziej stosowane są pozostałe dwa (aparaty 3 i 4). Aparaty 1 i 2 są w praktyce tym samym urządzeniem z różnym elementem mieszającym, odpowiednio koszyczkiem lub łopatką. Powyższe dwa aparaty składają się ze stanowisk pomiarowych umieszczonych w łaźni wodnej lub innym naczyniu grzewczym, ponieważ podczas badania musi być zapewniona temperatura odpowiednia dla danej postaci leku (37°C ± 0,5°C lub 32°C ± 0,5°C). Pojedyncze stanowisko zwykle zawiera:
przezroczystą, okrągłodenną zlewkę najczęściej o pojemności 1000 ml, wykonaną ze szkła lub innego obojętnego tworzywa, białego lub oranżowego (dla substancji fotolabilnych), zaopatrzoną w pokrywę chroniącą przed parowaniem, z otworami na mieszadło, termometr, kaniule lub inny element do pobierania próbek, np. pipetę, opcjonalnie otwór do wrzucenia tabletki,
element mieszający: wałek i cylindryczny koszyczek wykonany ze stali nierdzewnej (aparat 1) lub mieszadło łopatkowe (aparat 2) osadzone na wałku napędowym, które są wprawiane w ruch obrotowy o kontrolowanej szybkości.
Szczegółowe informacje o wymiarach czy kształcie powyższych elementów są zawarte w farmakopei. Poszczególne modele aparatów mogą różnić się między sobą akcesoriami i stopniem zautomatyzowania. Z tego względu mogą być wyposażone w dodatkowe elementy, m.in.: automatyczny system pobierania próbek i uzupełniania medium, pompę strzykawkową, filtr czy kolektor frakcji.
Wskazówki praktyczne
W zależności od zastosowanego elementu mieszającego należy zastosować inną szybkość mieszania: aparat koszyczkowy 100–150 obr./min; aparat łopatkowy 50–100 obr./min, w żadnym przypadku nie może jednak przekraczać 150 obr./min, w aparacie przepływowym szybkość przepływu płynu pomiędzy 4 ml/min a 50 ml/min.
Podczas badania żaden z elementów zestawu do przeprowadzania testu dostępności ani żadne urządzenie z otoczenia, w którym znajduje się aparat, nie może powodować ruchu, wibracji lub wstrząsów, które mogłyby zakłócić wyniki testu. Jedynym źródłem wibracji czy wstrząsów może być obracający się element mieszający.
W aparacie 1 postać leku umieszcza się w suchym koszyczku, następnie zanurza koszyczek w płynie i wprowadza w ruch obrotowy, natomiast w przypadku aparatu łopatkowego jednostkę postaci leku, np. tabletkę, umieszcza się na dnie zlewki pod łopatką i dopiero uruchamia mieszanie.
Użyte do badania jednostki postaci leku nie mogą być uszkodzone (np. tabletki nie mogą być ułamane, pokruszone).
Płyny akceptorowe
Do badania dostępności farmaceutycznej stosuje się z reguły roztwory wodne o odpowiednim składzie, pH i sile jonowej odzwierciedlające warunki, którym będzie podlegać postać leku po podaniu, np. warunki panujące w poszczególnych odcinkach przewodu pokarmowego (w żołądku na czczo pH 1–2, po posiłku pH 2‑5 lub w jelicie cienkim pH ok. 6‑8).
Głównymi składnikami płynów akceptorowych są substancje doprowadzające roztwór do odpowiedniego odczynu i siły jonowej. W specyficznych sytuacjach mogą jednak znajdować się w nich także inne związki, takie jak enzymy trawienne (np. pankreatyna, pepsyna), substancje obniżające napięcie powierzchniowe (np. laurylosiarczan sodu) lub inne składniki nieorganicznie i organiczne.
Wśród najpopularniejszych płynów można wymienić: kwas solny lub kwas solny z chlorkiem sodu, bufory: fosforanowy o pH 4,5 lub 6,8 oraz octanowy o pH 4,5, rzadziej wykorzystywane są np. bufory o innym pH czy też płyny „fizjologiczne”, np. sztuczny płyn żołądkowy lub jelitowy itp. Zarówno rodzaj płynu, jak i jego objętość należy dostosować do właściwości fizykochemicznych API, substancji pomocniczych i rodzaju badanej postaci leku. Podczas badania muszą być zapewnione warunki sink, to znaczy, że ilość uwolnionej substancji leczniczej nie może wpływać na dalszy proces uwalniania. Objętość medium musi być tak dobrana, aby po uwolnieniu z danej postaci leku maksymalnej ilości substancji czynnej jej stężenie było przynajmniej 3 do 10 razy mniejsze niż stężenie roztworu nasyconego. W aparatach 1 i 2 badanie najczęściej prowadzi się przy objętości płynów od 500 do 1000 ml (zwyczajowo 900 ml).
W przypadku substancji trudno rozpuszczalnych zapewnienie warunków sink może być trudne do osiągnięcia, z tego względu dopuszcza się użycie substancji powierzchniowo czynnych (np. laurylosiarczanu sodu, Tweenu 20–80, Brij). Surfaktanty dodaje się w najmniejszej koniecznej ilości, zwykle w stężeniu od 0,01 do 4,0%. Innym skutecznym rozwiązaniem w takich przypadkach może być przeprowadzenie testu w aparacie przepływowym. Nie powinno się natomiast prowadzić badań z wykorzystaniem roztworów wodno‑organicznych.
Wskazówki praktyczne
Temperatura płynów akceptorowych podczas odmierzania ich objętości powinna mieścić się w zakresie 20–25ºC.
Płyny do uwalniania muszą być ogrzane przed rozpoczęciem badania do temperatury odpowiedniej dla badanej postaci leku (w przypadku doustnych postaci leku do 37°C ± 0,5ºC), a następnie ich temperatura musi być kontrolowana i utrzymana na takim samym poziomie przez cały czas badania.
Podczas badania nie powinno być pęcherzyków powietrza w płynie akceptorowym, bo mogą one zaburzyć wynik badania. Z tego względu płyny, zwłaszcza przy stosowaniu aparatu przepływowego, należy odpowietrzyć przed zastosowaniem.
W przypadku kilkukrotnego pobierania prób objętość płynu akceptorowego zwykle uzupełnia się równoważną objętością świeżego medium, najlepiej ogrzanego do temperatury, w której prowadzone jest badanie. Jeżeli udowodniono, że można ten etap pominąć, należy wówczas w obliczeniach uwzględnić zmniejszającą się całkowitą objętość płynu.
Procedura wykonania badania uwalniania substancji czynnej ze stałych doustnych postaci leku
Dobór czasu badania, rodzaju medium i liczby punktów pobrań jest różny w zależności od postaci leku i celu badania.
Jeżeli badanie dostępności farmaceutycznej ma zostać wykorzystane do oceny powtarzalności poszczególnych serii produkcyjnych w rutynowych analizach kontrolnych, to wystarczy, aby test został przeprowadzony w jednym punkcie czasowym (dla postaci o niemodyfikowanym uwalnianiu) lub tylko w kilku punktach dla postaci o modyfikowanym lub opóźnionym uwalnianiu (tabela 1). Z kolei testy wielopunktowe, o kilku lub kilkunastu punktach pomiarowych, umożliwiają wykreślenie całego profilu uwalniania API z danego leku; na tej podstawie można wyznaczyć szybkość i stopień uwalniania substancji czynnej. Są przydatne podczas tworzenia nowych formulacji i wymagane podczas rejestracji leku.
Rodzaj medium | Czas testu | Ilość [%] substancji czynnej uwolniona w określonym czasie |
|---|---|---|
Tabletki o niemodyfikowanym uwalnianiu (szybko uwalniające) | ||
Jeden rodzaj | Zwykle 45 min lub krócej | 80% po czasie określonym w specyfikacji danego produktu, zwykle jeden punkt czasowy jest wystarczający, ale w niektórych przypadkach może być konieczne pobranie prób w kilku punktach czasowych |
Tabletki o modyfikowanym (przedłużonym) uwalnianiu | ||
Proces uwalniania najczęściej nie zależy od pH, dlatego test można prowadzić w jednym środowisku, np. buforze fosforanowym pH 6,8 | Najczęściej 6–8 godz., ale może być dłużej, nawet do 24 godz. Pobieranie prób w kilku punktach czasowych, przynajmniej trzech | Dla każdego punktu czasowego określona jest ilość (lub zakres) uwolnionej API wg specyfikacji produktu. Punkty pomiarów powinny być tak dobrane, aby umożliwiały sprawdzenie, czy:
|
Doustne postacie leku o opóźnionym uwalnianiu | ||
W zależności od zastosowanych warunków badania oraz budowy i składu leku sposób uwalniania substancji czynnej z takich postaci może być różny (np. porcjami lub całkowicie), dlatego wymagania dotyczące przeprowadzenia testu są ustalane indywidualnie dla każdego przypadku | ||
Tabletki dojelitowe jako przykład leku o opóźnionym uwalnianiu | ||
Dwa rodzaje mediów imitujące zmieniające się środowisko w przewodzie pokarmowym. Pierwszy punkt: kwas solny, drugi punkt: odpowiedni bufor (zaleca się bufor fosforanowy pH 6,8) | Przynajmniej dwa punkty czasowe. Pierwszy punkt: 1–2 godz., drugi punkt: najczęściej czas taki, jak dla tabletek o niemodyfikowanym uwalnianiu | Pierwszy punkt: określa górny limit uwolnionej substancji w środowisku kwasowym, zazwyczaj nie więcej niż 10%, drugi punkt: nie mniej niż 80% |
Wskazówki praktyczne
Próby powinny być pobierane w powtarzalny sposób, z głębokości odpowiadającej połowie wysokości między powierzchnią płynu w zlewce a górną krawędzią łopatki lub koszyczka, w odległości 1 cm od ścianki naczynia.
Próby powinny być pobierane w wyznaczonym czasie, a dopuszczalne odchylenie wynosi ±2%. Zatem przykładowo, jeżeli próba miała zostać pobrana w punkcie czasowym wynoszącym 20 min, to powinno się ją pobrać w przedziale czasu od 19 min 36 s do 20 min 24 s.
Próby należy sączyć bezpośrednio podczas pobierania płynu z naczynia pomiarowego (np. przez kaniulę z filtrem) lub zaraz po pobraniu (np. przez sączek strzykawkowy), aby pozbyć się nierozpuszczonych fragmentów postaci leku, z których mogłaby uwalniać się substancja czynna. Chyba, że wykazano, że sączenie nie jest konieczne. W przeciwnym razie istnieje ryzyko otrzymania zawyżonego wyniku oznaczania. Najlepiej, aby ze względu na ryzyko wykrystalizowania związku medium nie uległo schłodzeniu podczas sączenia. Zastosowany sączek nie może adsorbować na swojej powierzchni uwalnianej substancji ani zawierać ekstrahujących się związków.
Termometr należy usunąć z naczynia pomiarowego na czas badania, chyba że udowodniono brak wpływu jego obecności na wyniki analizy.
Podczas testów kilkupunktowych elementy pobierające, np. kaniule z filtrami zewnętrznymi, powinno się wyjmować z płynu akceptorowego po pobraniu prób, ponieważ każdy dodatkowy element w zlewce może zaburzać hydrodynamikę naczynia i wpływać na szybkość uwalniania.
Analiza ilościowa uwolnionej API
Po pobraniu i przesączeniu próby należy oznaczyć w niej zawartość substancji czynnej (lub w przypadku preparatów – złożonych substancji czynnych). Do tego celu najczęściej wykorzystuje się wysokosprawną chromatografię cieczową (HPLC) lub spektrofotometrię UV–VIS. Metoda musi charakteryzować się selektywnością wobec oznaczanych substancji czynnych i gwarantować otrzymanie precyzyjnych i dokładnych wyników.
Interpretacja wyników badania uwalniania substancji czynnej ze stałych doustnych postaci leku
W przypadku testów jednopunktowych określa się wymaganą ilość API (Q), która uwalnia się do medium w określonym czasie. Powyższą wartość wyraża się jako procent wartości deklarowanej przez producenta. Wynik porównuje się z przyjętymi w farmakopei kryteriami akceptacji.
Dla stałych postaci leku FP XII opisuje możliwość wykonania badania na trzech poziomach, jeżeli nie zostały spełnione wymagania na wcześniejszym poziomie akceptacji. W pierwszym etapie badanie wykonuje się dla 6 jednostek, a dopiero w przypadku niespełnienia kryteriów akceptacji dla dalszych 6 i ewentualnie kolejnych 12 jednostek (jednostką może być np. tabletka, kapsułka). Dla danego rodzaju doustnej stałej postaci leku FP XII podaje odpowiednie dopuszczalne odchylenia od przyjętych norm na poszczególnych poziomach, przy czym interpretacja wyników dotyczy odpowiednio 6, 12 lub 24 jednostek.
Przykładowo, jeżeli w badaniu na poziomie S1 dla każdej z 6 jednostek danej postaci leku o niemodyfikowanym uwalnianiu otrzymano wynik nie mniejszy niż wartość Q + 5% (wartość Q wynosi 75%), to uznaje się, że lek spełnia kryterium akceptacji. W przeciwnym wypadku należy powtórzyć badanie dla kolejnych 6 jednostek leku (poziom S2). Przy czym średnia wartość uzyskana dla 12 jednostek (S1 + S2) nie może być mniejsza niż Q, a dla wszystkich jednostek nie mniejsza niż Q - 15% (czyli 60%). W przypadku niespełnienia kryterium akceptacji z poziomu S2 wykonuje się badanie dla kolejnych 12 jednostek. Średnia wartość uzyskana dla 24 jednostek (S1 + S2 + S3) nie może być mniejsza niż Q, najwyżej 2 jednostki mogą mieć wartość mniejszą niż Q - 15% (czyli 60%), jednak dla wszystkich jednostek otrzymana wartość uwolnionej substancji nie może być mniejsza niż Q - 25% (czyli 50%).
Dla postaci o przedłużonym lub opóźnionym uwalnianiu FP XII przyjmuje odpowiednio inną interpretację wyników.
Określenie profilu uwalniania leku
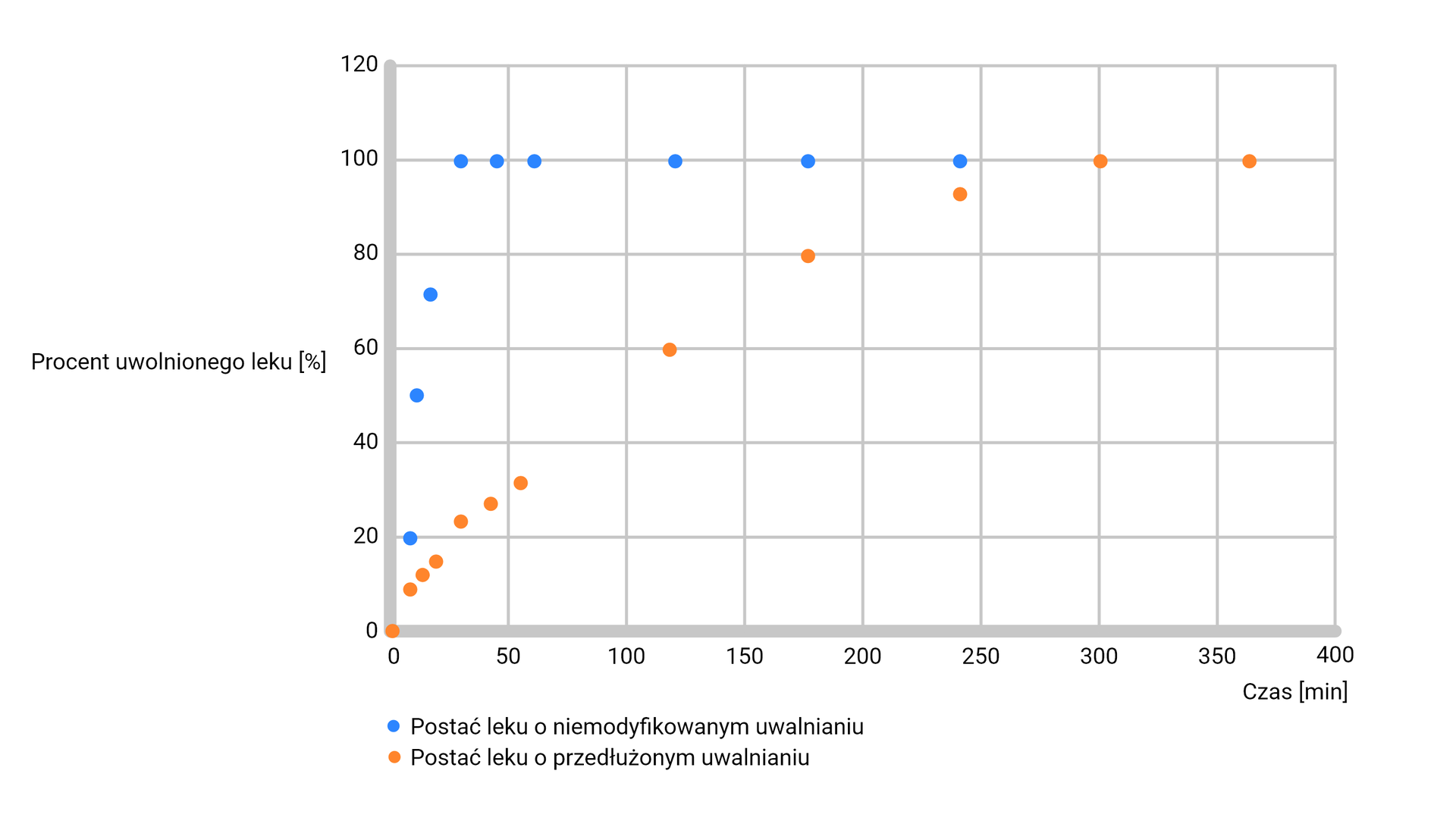
Wyniki badania uwalniania substancji czynnej z postaci leku umożliwiają także określenie profilu uwalniania leku. W tym celu na podstawie stężenia leku w płynie akceptorowym w każdej z pobranych próbek w określonych przedziałach czasowych należy wykreślić krzywą zależności ilości uwolnionej substancji leczniczej (najczęściej wyrażonej jako procent całkowitej dawki deklarowanej) od czasu. Na rycinie 1 zaprezentowano przykładowy wykres przedstawiający profil uwalniania leku z postaci leku o niemodyfikowanym uwalnianiu (szybko uwalniającej substancję aktywną) oraz z postaci o przedłużonym uwalnianiu.
Kroki postępowania w symulatorze
Czynności wykonywane przed rozpoczęciem badania:
Włączanie urządzenia
Napełnianie łaźni wodnej
Umiejscowienie naczyń pomiarowych (zlewek okrągłodennych)
Napełnienie naczyń pomiarowych medium
Ustawianie temperatury łaźni wodnej
Mocowanie mieszadeł
Unoszenie/opuszczanie panelu mieszadeł
Ustawianie szybkości obrotowej mieszadeł
Badanie uwalniania substancji leczniczej z tabletek zwykłych do 0,1 mol/l HCl o pH = 1 (medium):
Umieszczanie tabletek w naczyniu pomiarowym
Rozpoczęcie badania
Pobieranie próbek do badań
Uzupełnienie naczyń pomiarowych medium
Zakończenie badania
Czynności wykonywane po zakończeniu badania:
Opróżnianie naczyń pomiarowych
Usuwanie wody z łaźni i mycie zbiornika
Wyłączanie urządzenia

Zasób interaktywny dostępny pod adresem https://zpe.gov.pl/a/DQbEt8lMH