Przeczytaj
Rodzaje badań diagnostycznych płodu
- Nazwa kategorii: Rodzaje badań diagnostycznych płodu
- Nazwa kategorii: ultrasonografia
- Nazwa kategorii: amniopunkcja
- Nazwa kategorii: analiza DNA
- Nazwa kategorii: cytogenetyczne [br] badania [br] prenatalne Koniec elementów należących do kategorii Rodzaje badań diagnostycznych płodu
- Elementy należące do kategorii Rodzaje badań diagnostycznych płodu
Najpowszechniej stosowaną metodą badania płodu jest ultrasonografia, niepowodująca ryzyka jego uszkodzenia. W obrazie USG doświadczony lekarz może dostrzec zewnętrzne anomalie płodu, jednak nie jest w stanie stwierdzić nieprawidłowości w obrębie DNA.
Informacji na temat zaburzeń DNA dostarczają badania biochemiczne i genetyczne. Można je przeprowadzić dzięki amniopunkcji (amniocentezie), polegającej na nakłuciu u ciężarnej jamy owodni i pobraniu płynu owodniowego, który otacza dziecko. Gdy płód rośnie, jego naskórek w naturalny sposób się złuszcza, w wyniku czego jego komórki pojawiają się w płynie owodniowympłynie owodniowym. Po ich pobraniu i hodowli poddaje się je badaniom laboratoryjnym. W trakcie podziałów komórek płodu oceniana jest liczba chromosomów, wyraźnie wówczas widocznych, co pozwala na stwierdzenie ewentualnych mutacji chromosomowych liczbowych lub na ich wykluczenie. Analiza składu płynu owodniowego daje jeszcze jedną ważną informację: znajduje się w nim tzw. białko płodowe, którego zbyt niski poziom wskazuje na występowanie u dziecka zespołu Downa.
Stwierdzenie niektórych chorób genetycznych dziecka, m.in. mukowiscydozymukowiscydozy, hemofiliihemofilii, anemii sierpowatej i fenyloketonuriifenyloketonurii, wymaga pobrania innych komórek niż naskórka płodu. Najczęściej badane są jego krwinki, które przedostały się do krwiobiegu matki. Analiza DNA krwinek płodu przy wykorzystaniu nowoczesnych technik biologii molekularnej pozwala wykryć ewentualne mutacje genowe. Rozpoznanie wad genomu dziecka daje szansę podjęcia odpowiedniego leczenia na wczesnym etapie.
Gdy zachodzi potrzeba potwierdzenia bądź wykluczenia zmiany struktury lub liczby chromosomów, wykonywane są prenatalne badania cytogenetyczne. Przeprowadza się je przede wszystkim w przypadku istnienia czynników ryzyka wystąpienia wad chromosomalnych, takich jak: wiek matki, niepowodzenia rozrodu, nieprawidłowe wyniki standardowych badań prenatalnych (np. USG), zaburzenia chromosomowe stwierdzone u jednego z rodziców. Podczas cytogenetycznego badania prenatalnego pobierana jest próbka z tkanek płodu.
Zmiany liczby chromosomów w kariotypie człowieka
Choroby spowodowane zmianą liczby autosomów
Analiza kariotypu płodu pozwala na wykrycie zmian w liczbie chromosomów.
Chromosomy komórek diploidalnych tworzą pary homologiczne, podobne do siebie pod względem kształtu i zawartości genetycznej. Jeśli w jednej z takich par pojawi się nadliczbowy chromosom (czyli zamiast dwóch w parze są trzy), występuje tzw. trisomia. Jeśli natomiast w parze brakuje jednego chromosomu, taką zmianę określa się jako monosomię. Trisomia i monosomia to przykłady mutacji aneuploidalnych – mutacji liczbowych dotyczących pojedynczych chromosomów.
Najczęściej występującą w populacji ludzkiej mutacją aneuploidalną jest trisomia 21 pary chromosomów. Mimo że ten mały chromosom zawiera stosunkowo niewiele genów, obecność w genomie jego dodatkowej kopii powoduje zmiany chorobowe określane jako zespół Downa. Do najbardziej typowych objawów tego schorzenia należą: niedorozwój umysłowy o różnym nasileniu, niski wzrost, zaburzone proporcje ciała oraz liczne zmiany anatomiczne i fizjologiczne, dotyczące zwłaszcza układu krwionośnego. Kariotyp osoby z tą mutacją to: 47,XX,+21 lub 47,XY,+21.
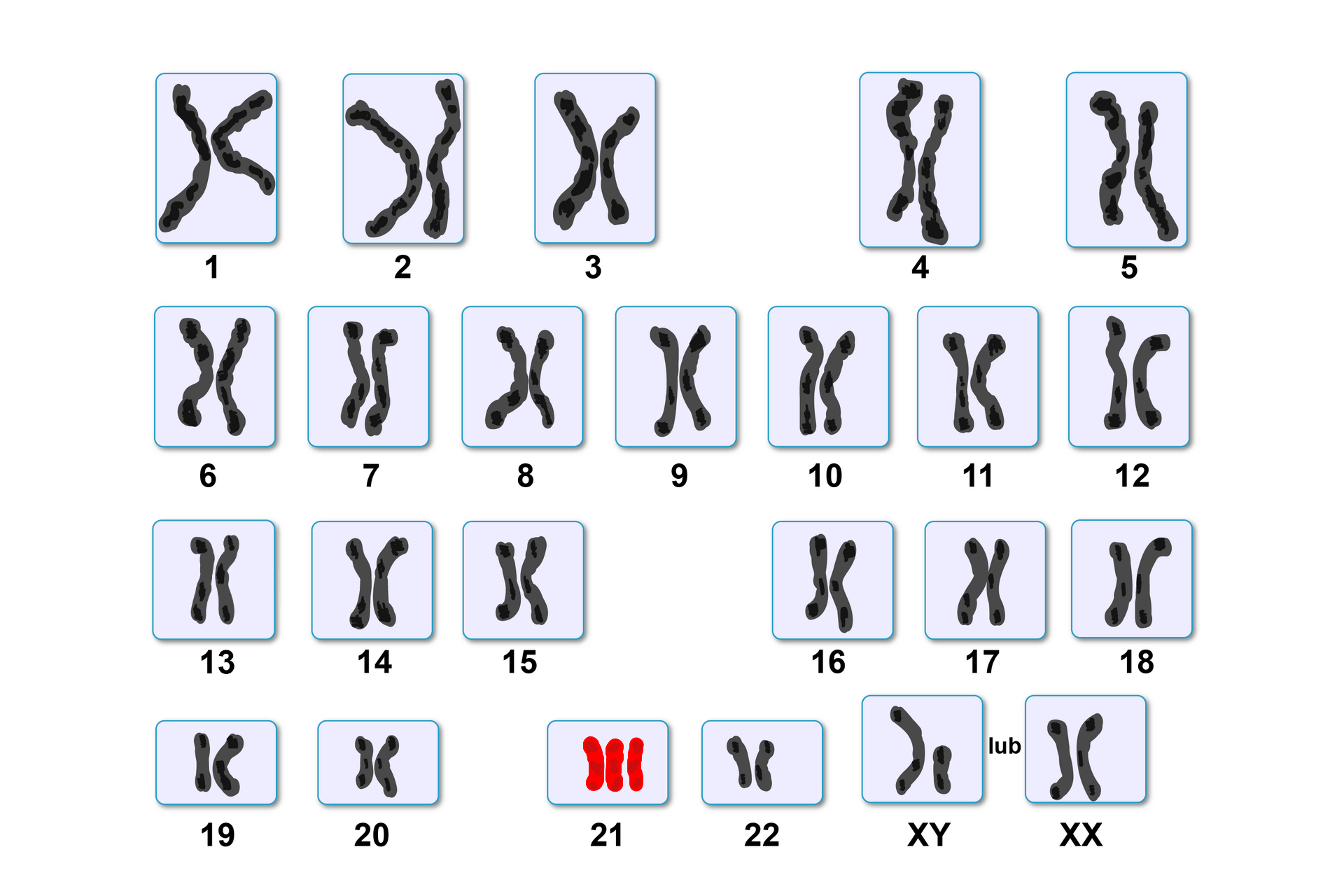
Istnieje związek między wiekiem matki a ryzykiem wystąpienia zespołu Downa u potomstwa: 1 dziecko na 30 urodzonych przez kobiety po 40. roku życia choruje na zespół Downa. Przypuszcza się, że może to być spowodowane faktem, że późno dojrzewająca komórka jajowa jest znacznie bardziej narażona na uszkodzenia, np. przez czynniki mutagenne. Po 35. roku życia częściej występują również zaburzenia w podziale mejotycznym, prowadzącym do powstania komórek jajowych. Chromosomy rozchodzą się wtedy nieprawidłowo i istnieje ryzyko, że komórka jajowa zamiast jednego chromosomu z 21 pary otrzyma oba. Jeśli zapłodni ją zdrowy plemnik, niosący po jednym chromosomie z każdej pary, również z 21, powstanie organizm trisomiczny.
Do poważnych zmian rozwojowych należy także trisomia chromosomu 18, która odpowiada za zespół Edwardsa. Charakteryzuje się on m.in. małogłowiem, nieprawidłowościami w budowie przepony, rozszczepem wargi lub podniebienia i zniekształceniami w obrębie twarzoczaszki. Przeprowadzenie badań pozwala na odróżnienie trisomii chromosomu 18 od innych chorób o podłożu genetycznym, które mogą dawać podobne objawy. Jest to ważne, ponieważ dzieci cierpiące na zespół Edwardsa muszą być karmione przez specjalistyczne sondy z powodu braku odruchu ssania. Kariotyp osoby z tą mutacją to: 47,XX,+18 lub 47,XY,+18.

Natomiast trisomia chromosomu 13 związana jest z zespołem Pataua. Do jego objawów należą: wady w obrębie narządu wzroku (często cyklopia, czyli jednoocze), a także wady układu krążenia, nerek, zaburzenia w budowie kończyn czy w rozwoju układu nerwowego. Wystąpienie zespołu Pataua zwiększa ryzyko poronienia, a przeżywalność dzieci do pierwszego roku życia jest niezwykle mała. Analiza kariotypu w tym przypadku ma ogromne znaczenie, gdyż istnieje korelacja między wiekiem matki a częstością wystąpienia zespołu Pataua. Kariotyp osoby z tą mutacją to: 47,XX,+13 lub 47,XY,+13.

Choroby spowodowane zmianą liczby chromosomów płci
Skutkiem nieprawidłowego rozejścia się chromosomów w trakcie mejozy mogą być również mutacje dotyczące liczby chromosomów płci. U kobiet dochodzi do monosomii chromosomów X (X0) – zamiast dwóch występuje tylko jeden. Konsekwencją braku jednego chromosomu X jest zespół Turnera, który objawia się obniżoną inteligencją, bezpłodnością, niskim wzrostem, wadami układu krwionośnego i moczowego. Kariotyp kobiety z tą mutacją to: 45,X.

Natomiast u kobiet mających dodatkowy, trzeci chromosom X (trisomia XXX) nie występują jednoznaczne objawy. U 25% kobiet z trisomią XXX pojawiają się problemy z płodnością, ponadprzeciętny wzrost oraz nieco niższy od przeciętnego poziom rozwoju umysłowego. Kariotyp osoby z tą mutacją to: 47,XXX.
U mężczyzn jedną z najczęstszych chorób wywoływanych nieprawidłową liczbą chromosomów płci jest zespół Klinefeltera, spowodowany obecnością dodatkowego chromosomu X w kariotypie męskim (trisomia XXY). Efektami tej mutacji są kobieca sylwetka oraz powodujący bezpłodność niedorozwój jąder, jednak nie występuje obniżenie inteligencji. Kariotyp mężczyzny z tą mutacją to: 47,XXY.

U mężczyzn możliwa jest też trisomia chromosomów płci (XYY), która nie powoduje widocznych zmian chorobowych. Przez pewien czas uważano, że dodatkowy chromosom Y jest źródłem zwiększonej agresji u mężczyzn, ale ostatnie badania nie potwierdzają tej hipotezy.
Zmiany struktury chromosomów w kariotypie człowieka
Analiza kariotypu płodu połączona z technikami znakowania DNA pozwala na dokładną ocenę struktury chromosomów. Dzięki temu możliwe jest stwierdzenie wystąpienia translokacjitranslokacji między chromosomami. Jedną z tego typu nieprawidłowości jest chromosom Philadelphia, związany z obecnością przewlekłej białaczki szpikowej. Powstaje on na skutek translokacji zachodzącej między chromosomem 9 pary i chromosomem 22 pary.
W leczeniu przewlekłej białaczki szpikowej duże znaczenie ma szybkość rozpoznania choroby. W początkowym okresie (kilku lat) choroba przebiega powoli, jednak wraz z upływem czasu wzrasta ilość białych krwinek, dlatego brak skutecznego leczenia w początkowej fazie powoduje groźne powikłania, prowadzące do śmierci chorego.

Translokacja fragmentu chromosomu 22 i chromosomu 11 powoduje 90% przypadków mięsaka Ewinga – złośliwego nowotworu kości i tkanek miękkich, najczęściej występującego u dzieci i młodzieży.
Znacznie rzadziej stwierdza się inne translokacje. Na podstawie wyników badań genetycznych ustalane jest ostateczne rozpoznanie choroby. Umożliwia to wdrożenie odpowiednich metod terapii celowanej, skuteczniejszej od metod standardowych. Szacuje się, że dzięki zastosowaniu leczenia celowanego można uratować nawet 8 na 10 chorych.

Delecja krótkiego ramienia chromosomu 5 powoduje wystąpienie zespołu cri du chat (inaczej zespołu kociego krzyku), którego charakterystycznym objawem jest nietypowy, piskliwy płacz u dziecka, przypominający miauczenie kota.
Prawdopodobieństwo wystąpienia zespołu cri du chat nie przekracza 1%, jednak wzrasta ono do 15% w przypadku, gdy u jednego z rodziców stwierdza się translokację fragmentu chromosomu 5.

Analiza kariotypu umożliwia także wykrycie zmiany w postaci tzw. chromosomu pierścieniowego. Powstaje on na skutek połączenia się dwóch ramion chromosomu w pierścień. W efekcie występuje szereg nieprawidłowości morfologicznych i fizjologicznych, obejmujących zmiany w budowie czaszki, niski wzrost, opóźnienie umysłowe czy niedoczynność tarczycy i przytarczyc. Chociaż chromosomy pierścieniowe są bardzo rzadkie, mogą dotyczyć każdego ludzkiego chromosomu: autosomów lub chromosomu X.
Objawy i nasilenie nieprawidłowości w funkcjonowaniu organizmu u osób z chromosomem pierścieniowym zależą od ilości utraconej informacji genetycznej oraz jej lokalizacji. W sytuacji, gdy końce chromosomu łączą się bez utraty materiału genetycznego, nie występują poważne zmiany morfologiczne ani fizjologiczne. Natomiast gdy dojdzie do delecji, powstają poważne zaburzenia związane z konkretnym chromosomem. Leczenie w przypadku chromosomu pierścieniowego 15 polega jedynie na niwelowaniu negatywnych objawów.

Więcej informacji na temat chorób genetycznych spowodowanych abberacjami chromosomowymi strukturalnymi znajdziesz w e‑materiale:Choroby człowieka związane ze zmianą struktury chromosomów.
Poradnictwo genetyczne
Analiza kariotypu osób starających się o dziecko pozwala na określenie występowania wad genetycznych bądź zespołu aberracji chromosomowych, które mogą być dziedziczone lub przyczyniać się do niepłodności lub nawet bezpłodności pary. Osoba, u której wykazano tego typu wadę, otrzymuje informacje o dziedziczeniu zmiany w kariotypie (prawdopodobieństwie urodzenia zdrowego potomka) oraz o możliwości leczenia lub innego rodzaju pomocy i wsparcia.
Czynnikiem przemawiającym za wykonaniem badania kariotypu jest m.in. wystąpienie chorób genetycznych w rodzinie pary starającej się o potomstwo. Szacuje się, że aberracje chromosomowe dotyczą ok. 0,6% populacji ogólnej, jednak wśród bezpłodnych mężczyzn wartość ta rośnie do 14%. Analiza kariotypu u niemal 15 tys. par w Chinach wykazała, że nieprawidłowy kariotyp występował wśród 6,84% mężczyzn i 0,84% kobiet.
W przypadku par, u których występuje problem z poczęciem dziecka, analiza kariotypu pozwala wykluczyć genetyczne przyczyny bezpłodności, a w przypadku wykrycia nieprawidłowości – pozwala określić, czy problem leży po stronie mężczyzny czy kobiety. Dzięki temu możliwe jest powzięcie odpowiednich działań w dalszych staraniach o potomstwo.
Aberracje chromosomowe w badaniu kariotypu stwierdza się u 13% mężczyzn, u których występuje azoospermia – brak plemników w nasieniu. Z azoospermią oraz oligozoospermią (obniżoną liczbą plemników w nasieniu) powiązano też mikrodelecje w obrębie chromosomu Y, odpowiadającego za proces powstawania plemników.
Istotna jest analiza kariotypu u kobiet, które starają się o dziecko po przekroczeniu 35 roku życia, kiedy wzrasta prawdopodobieństwo wystąpienia ciężkich wad genetycznych u płodu.
Wskazaniami do przeprowadzenia badań genetycznych są więc:
wiek przyszłej matki powyżej 35 roku życia;
urodzenie w przeszłości dziecka z chorobą metaboliczną lub wadą wrodzoną;
anomalie chromosomowe płodu lub aberracje chromosomów płciowych w poprzednich ciążach;
nieprawidłowy wynik USG przesiewowego wykonanego między 11 a 14 tygodniem ciąży;
wada ośrodkowego układu nerwowego u płodu;
wady chromosomowe u rodziców;
niepowodzenia w rozrodzie;
wystąpienie nowotworów;
potwierdzone przypadki urodzeń dzieci z wadami genetycznymi w rodzinie przyszłej matki lub przyszłego ojca;
korzystanie z techniki wspomaganego rozrodu, jaką jest zapłodnienie in vitro.
W ocenie ryzyka istnienia aberracji chromosomowych skutkujących obniżeniem płodności ważne jest również badanie częstotliwości ich występowania, a także zidentyfikowanie ewentualnych przyczyn środowiskowych zwiększających ryzyko pojawienia się nieprawidłowości w kariotypie gamet.
Słownik
pobranie próbki płynu owodniowego w celu przeprowadzenia badań prenatalnych
(ang. G‑banding by trypsin with Giemsa) podstawowe badanie cytogenetyczne, pozwalające uzyskać całościowy obraz aberracji chromosomowych w komórkach
metoda diagnostyki prenatalnej polegająca na przezbrzusznej lub przezszyjkowej biopsji kosmków kosmówki pod kontrolą USG
odcinek genu kodujący sekwencję aminokwasów wchodzącą w skład białka
wrodzone zaburzenie przemiany fenyloalaniny (niezbędnego aminokwasu egzogennego), w którego wyniku dochodzi do uszkodzenia układu nerwowego
(ang. fluorescence in situ hybridization) technika cytochemiczna; polega na hybrydyzacji fragmentu DNA lub RNA ze specyficznymi sondami znakowanymi barwnikami fluorescencyjnymi
gen znajdujący się na długim ramieniu chromosomu 7 w locus 7q31.2; koduje białko CFTR (ang. cystic fibrosis transmembrane conductance regulator – błonowy regulator przewodnictwa), tworzące kanał chlorkowy w błonie komórkowej; mutacja tego genu wywołuje mukowiscydozę
dziedziczna skaza krwotoczna spowodowana niedoborem lub zaburzoną funkcją swoistych białek osocza, niezbędnych do prawidłowego krzepnięcia krwi
nakłucie pępowiny płodu przy badaniach prenatalnych
schorzenie dziedziczne (dziedziczone autosomalnie recesywnie) cechujące się nieprawidłową funkcją wszystkich gruczołów wydzielania zewnętrznego oraz zaburzeniami gospodarki jonowej, w szczególności wydzielania jonów chlorkowych na zewnątrz komórek nabłonkowych
płyn znajdujący się w jamie owodni, otaczający zarodek, a następnie płód w okresie życia wewnątrzmacicznego
wyznakowany fragment DNA lub RNA, wykorzystywany do lokalizowania komplementarnych sekwencji DNA lub RNA
rodzaj aberracji chromosomowej, częsty podczas transformacji nowotworowej; polega na wzajemnym lub niewzajemnym przemieszczeniu się fragmentów DNA pomiędzy dwoma niehomologicznymi chromosomami