Przeczytaj
Choroby genetyczne człowieka spowodowane są mutacjamimutacjami zachodzącymi w strukturze DNA. W zależności od zakresu mutacji wyróżnia się choroby:
jednogenowe – wywołane mutacjami w obrębie jednego genu;
wielogenowe – wywołane mutacjami w obrębie różnych genów i indukowane czynnikami środowiska;
chromosomalne – wywołane mutacjami w strukturze lub liczbie chromosomów.
Więcej na temat mutacji przeczytasz w e‑materiale: Mutacje – kryteria podziału i rodzajeMutacje – kryteria podziału i rodzaje.
Choroby jednogenowe
Choroby jednogenowe są w większości spowodowane mutacjami genów zlokalizowanych w jądrowym DNA. Dzieli się na dwie grupy: autosomalne i allosomalne.
Choroba autosomalna związana jest z mutacjami genu położonego na chromosomie autosomalnymchromosomie autosomalnym.
Choroba allosomalna wynika z mutacji genu położonego na chromosomie allosomalnymchromosomie allosomalnym.
Ze względu na sposób dziedziczenia wyróżnia się choroby jednogenowe: dziedziczone recesywnie i dziedziczone dominująco. Nieliczne choroby jednogenowe są wywołane obecnością mutacji w genach położonych w mitochondrialnym DNA.
Choroby jednogenowe dziedziczone autosomalnie recesywnie
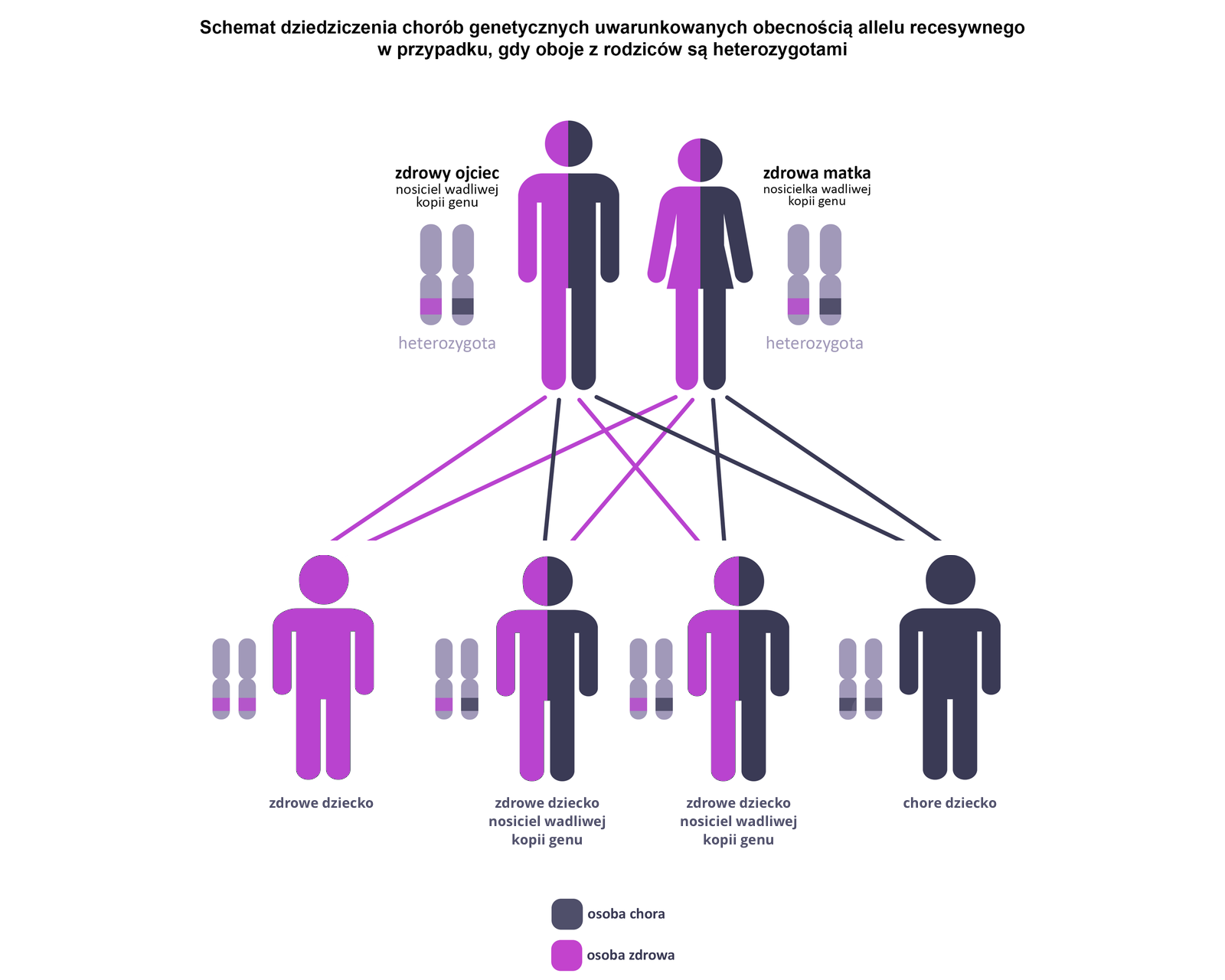
Choroby dziedziczone autosomalnie recesywnie najczęściej dotyczą genów kodujących białka enzymatyczne. Brak lub niedobór prawidłowej formy enzymu skutkuje zaburzeniami przemian metabolicznych organizmu. Do wystąpienia objawów choroby konieczna jest obecność dwóch wadliwych allelialleli (a) danego genu. Choroba ujawnia się u homozygot recesywnych (aa). HomozygotyHomozygoty dominujące (AA) i heterozygotyheterozygoty (Aa) nie wykazują objawów choroby – heterozygoty są jednak nosicielami wadliwego genu, który mogą przekazać potomstwu. Choroby jednogenowe dziedziczone autosomalnie recesywnie występują z jednakową częstością u obu płci.
Albinizm to choroba związana z zaburzeniami w procesie syntezy melanin. Przyczyną albinizmu są mutacje genu TYR kodującego tyrozynazę – enzym katalizujący reakcję przekształcania tyrozyny do 3,4‑dihydroksyfenyloalaniny, związku wykorzystywanego do syntezy melanin. Skutkiem mutacji jest niedobór lub brak barwnika w skórze, włosach i tęczówce oka. Choroba objawia się bardzo jasną skórą, białymi włosami i różowymi lub niebieskimi tęczówkami oczu. Dodatkowo u wielu albinotycznych osobników występuje oczopląs, światłowstręt i zmniejszona ostrość wzroku. Ludzie cierpiący na albinizm wykazują nadmierną wrażliwość na promieniowanie słoneczne, co zwiększa ryzyko poparzeń słonecznych i nowotworów skóry.

Alkaptonuria to choroba związana z zaburzeniami przemian metabolicznych kwasu homogentyzynowego będącego metabolitem pośrednim przekształceń tyrozyny i fenyloalaniny. Najczęstszą przyczyną choroby są mutacje genu HGD kodującego białko – oksygenazę homogentyzynianową (HGD). Białko HGD jest enzymem katalizującym przekształcenie kwasu homogentyzynowego do produktów cyklu Krebsa. Skutkiem mutacji jest niedobór lub brak białka HGD, czego konsekwencją jest gromadzenie się we krwi i płynach ustrojowych nadmiaru kwasu homogentyzynowego i produktów alternatywnego przekształcania tego związku. Nadmiar kwasu homogentyzynowego ma szkodliwy wpływ na tkankę chrzęstną. Choroba objawia się we wczesnym dzieciństwie niebiesko zabarwionym moczem. W późniejszym czasie dochodzi do rozwoju stanów zapalnych i zmian zwyrodnieniowych stawów, co przejawia się bólem i sztywnością stawów oraz trudnościami w poruszaniu się.
Anemia sierpowata to choroba związana z zaburzeniem syntezy hemoglobiny. Przyczyną choroby jest mutacja genu HBB kodującego łańcuch beta hemoglobiny. Mutacja punktowa jednego nukleotydu (tyminowego na adeninowy) w genie kodującym beta hemoglobinę skutkuje zmianą jednego aminokwasu – kwasu glutaminowego – na walinę, czego konsekwencją jest powstanie hemoglobiny S. Skutkiem mutacji jest obecność nieprawidłowej hemoglobiny S, która agreguje w erytrocytach, powodując zmianę ich struktury. Choroba objawia się niedokrwistością i niedotleniem tkanek organizmu. Niedokrwistość, czyli zmniejszona ilość hemoglobiny w organizmie, wynika z rozkładu sierpowatych erytrocytów w śledzionie. Niedotlenienie, zmniejszona podaż tlenu do komórek ciała, jest skutkiem mniejszego powinowactwa do tlenu nieprawidłowej hemoglobiny S w porównaniu do prawidłowej hemoglobiny A.

Mukowiscydoza to choroba związana z zaburzeniem transportu jonów chlorkowych przez błony komórkowe. Wśród populacji ludzi ras czarnej i żółtej występuje znacznie rzadziej. Przyczyną choroby są mutacje genu CFTR kodującego białko – przezbłonowy regulator transportu jonów (CFTR)przezbłonowy regulator transportu jonów (CFTR). Białko CFTR występuje na powierzchni komórek tkanki nabłonkowej dróg oddechowych, układu pokarmowego, trzustki, wątroby i gruczołów potowych. Skutkiem mutacji mogą być: brak białka CFTR lub zmniejszenie jego stabilności, a także zaburzenia regulacji jego funkcji. Choroba objawia się upośledzeniem transportu jonów chlorkowych oraz zwiększeniem absorpcji jonów sodowych i wody, w wyniku czego powstaje gęsty śluz. Obecność nadmiernych ilości śluzu w drogach oddechowych sprzyja rozwojowi chorobotwórczych mikroorganizmów, prowadząc do częstych i trudnych w leczeniu infekcji. Konsekwencją zmian w narządach układu oddechowego jest niewydolność oddechowa. Z kolei śluz gromadzący się w przewodach wyprowadzających trzustki utrudnia odpływ soku trzustkowego, przyczyniając się do rozwoju stanów zapalnych narządu i jego zwłóknienia. Nieprawidłowe funkcjonowanie nabłonka jelit oraz upośledzenie funkcji trzustki powodują zaburzenia wchłaniania substancji pokarmowych. W przypadku gruczołów potowych obserwuje się nadmierne wydzielanie wraz z potem jonów chlorkowych i sodowych. U mężczyzn choroba prowadzi do niedrożności lub zaniku nasieniowodów i w konsekwencji do bezpłodności.

Choroby jednogenowe dziedziczone autosomalnie dominująco
Choroby dziedziczone autosomalnie dominująco występują w ludzkiej populacji znacznie rzadziej niż choroby dziedziczone autosomalnie recesywnie. Do wystąpienia objawów choroby wystarcza tylko jeden wadliwy allel (A) danego genu. Choroba ujawnia się u heterozygotheterozygot (Aa) i homozygothomozygot dominujących (AA), przy czym homozygoty dominujące wykazują cięższy przebieg choroby lub mutacja jest dla nich letalnaletalna. Choroby jednogenowe dziedziczone autosomalnie dominująco występują z jednakową częstością u obu płci.

Choroba | Typ dziedziczenia | Częstość występowania | Charakter zaburzeń |
|---|---|---|---|
Albinizm | recesywny | 1:35 000 żywych urodzeń | zaburzenie syntezy |
Fenyloketonuria | recesywny | 1:10 000 żywych urodzeń u ludzi rasy białej | blok metaboliczny |
Alkaptonuria | recesywny | 1:100 000 żywych urodzeń u ludzi rasy białej | blok metaboliczny |
Anemia sierpowata | recesywny | 1:600 000 żywych urodzeń u ludzi rasy białej i 1:625 żywych urodzeń u ludzi rasy czarnej | zaburzenie syntezy |
Galaktozemia | recesywny | 1:23 000–1:40 000 żywych urodzeń u ludzi rasy białej | blok metaboliczny |
Mukowiscydoza | recesywny | 1:2000–1:4000 żywych urodzeń u ludzi rasy białej | zaburzenie przepływu jonów |
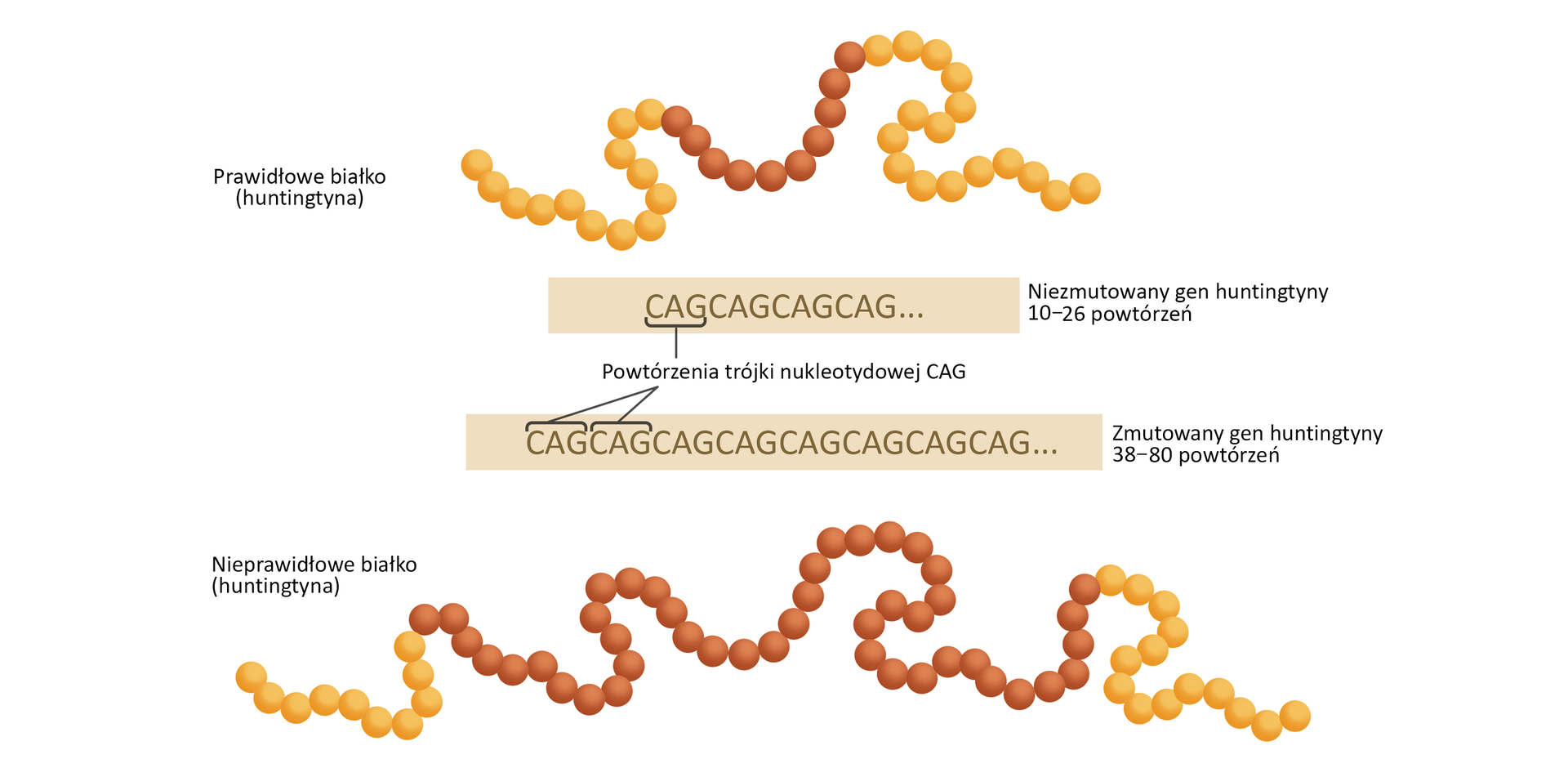
Pląsawica Huntingtona | dominujący | 4:100 000–7:100 000 żywych urodzeń | zaburzenie przewodnictwa nerwowego |
Choroby wielogenowe
Choroby wielogenowe są spowodowane mutacjami wielu genów zajmujących miejsca na różnych chromosomach. Współdziałanie produktów tych genów decyduje o ewentualnym wystąpieniu oraz natężeniu objawów. Pojedyncza mutacja nie przesądza więc o wystąpieniu konkretnej choroby, a dziedziczona jest jedynie pewna skłonność do jej rozwoju. To, czy obecność wadliwych genów ujawni się w postaci danej jednostki chorobowej, zależy od interakcji genów, prowadzonego trybu życia i wpływu czynników środowiska. Z tego względu choroby te określa się także mianem chorób wieloczynnikowych. Zalicza się do nich m.in. nadciśnienie, cukrzycę i schizofrenię.
Cechy wielogenowe są wypadkową działania produktów wielu genów, niesprzężonych ze sobą. Przykładem takiej cechy jest pigmentacja skóry człowieka, która zależy od 10–20 genów. Większość z nich koduje białka szlaku syntezy malaniny, czyli barwnika występującego w naskórku.
Słownik
(gr. allos – inny) forma genu; jedna z wersji genu różniąca się od pozostałych sekwencją nukleotydów; zajmuje określone miejsce na chromosomie
inaczej allosomy (gr. allos – inny, sṓma – ciało), chromosomy płci, heterochromosomy; chromosomy zawierające geny determinujące płeć osobnika
autosomy (gr. autós – sam, sṓma – ciało); wszystkie chromosomy wchodzące w skład kariotypu poza chromosomami płci; u organizmów diploidalnych układają się w pary chromosomów homologicznych (człowiek ma 22 pary chromosomów autosomalnych)
(ang. cystic fibrosis transmembrane conductance regulator); białko, które tworzy kanał chlorkowy w błonie komórkowej; kodowane przez gen CFTR znajdujący się na długim ramieniu chromosomu 7; nieprawidłowa forma tego białka wywołuje mukowiscydozę
aminokwas egzogenny, który musi być dostarczany do organizmu wraz z pokarmem; w organizmie zwierzęcym utlenia się do tyrozyny; choroba związana z zaburzeniami przemian metabolicznych fenyloalaniny to fenyloketonuria
cukier prosty należący do heksoz, wchodzi w skład laktozy; źródłem galaktozy jest laktoza (cukier mleczny) metabolizowana w organizmie człowieka do glukozy i galaktozy; w wyniku reakcji enzymatycznych galaktoza przekształcana jest do glukozy, u osób chorujących na galaktozemię przekształcenie to nie zachodzi
(gr. héteros – inny, zygōtós – połączony) organizm diploidalny zawierający dwa różne allele danego genu (Aa)
(gr. homós – taki sam, zygōtós – połączony) organizm diploidalny zawierający dwa jednakowe allele danego genu (AA –homozygota dominująca i aa – homozygota recesywna)
białko występujące w komórkach nerwowych, uczestniczące w przekazywaniu impulsów nerwowych oraz regulujące przebieg procesów transkrypcji, translacji i transportu komórkowego; kodowane przez gen IT15 leżący na krótkim ramieniu chromosomu 4
(łac. mutatio – zmiana) nagła, losowa, skokowa zmiana w materiale genetycznym powstała samoistnie lub na skutek działania czynników mutagennych
mutacja wywołująca poważne wady genetyczne, które w znacznym stopniu upośledzają funkcjonowanie organizmu i w konsekwencji prowadzą do śmierci
aminokwas egzogenny; powstaje z egzogennej dla zwierząt fenyloalaniny; u kręgowców z tyrozyny powstaje wiele ważnych biologicznie związków: hormony (adrenalina, tyroksyna), melaniny; w komórce, w obecności kwasu askorbinowego, tyrozyna rozpada się na kwasy: fumarowy i acetylooctowy, które następnie za pośrednictwem acetylokoenzymu A są włączane do cyklu Krebsa; zachwianie rozpadu tyrozyny prowadzi do ciężkiej choroby — alkaptonurii