Przeczytaj
Techniki mapowania genomów rozwijały się przez wiele lat. Choć obecnie pierwotne analizy wydają się być bardzo nieprecyzyjne, to wszelkie zdobyte informacje pozwoliły m.in. na ustalenie kolejności genów na chromosomach. Dane te okazały się nieocenione w momencie, w którym możliwe było sekwencjonowanie genomów. Dzięki poznanej kolejności genów łatwiejsze stało się dopasowanie sekwencjonowanych fragmentów w trakcie składania w całość sekwencji genomu ludzkiego.
Ustalenie pełnej sekwencji genomu pozwala na analizę genów związanych z chorobami czy badanie pokrewieństwa między organizmami. Poznanie sekwencji genomów innych organizmów umożliwia ustalenie ścieżki ewolucyjnej człowieka, poprzez ustalenie regionów niezmiennych ewolucyjnie. Analiza porównawcza tych regionów pozwala na odtworzenie prawdopodobnego drzewa filogenetycznego, prowadzącego do człowieka. Genomy mikroorganizmów służą np. w opracowaniu potencjalnych leków i szczepionek.
Mapy genomu
W celu scharakteryzowania genomugenomu ludzkiego tworzy się trzy rodzaje map: genetyczne, cytogenetyczne i fizyczne.
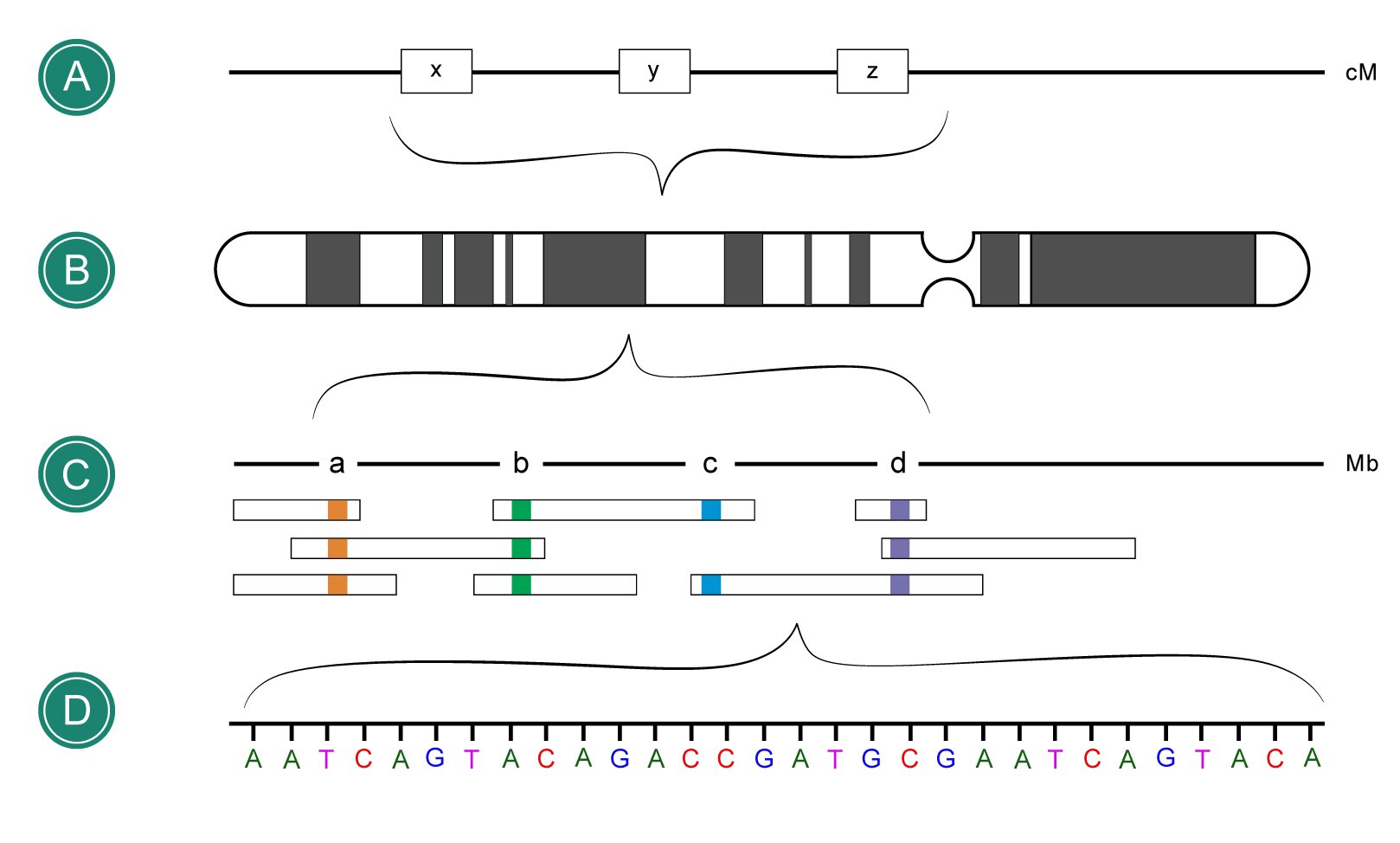
Mapa genetyczna – graficzne przedstawienie uporządkowania genów w cząsteczce DNA. Obejmuje zarówno kolejność położenia genów, jak i odległości między nimi. Mapy genetyczne mogą być liniowe lub koliste, w zależności od struktury odwzorowywanej cząsteczki DNA. Jednostką opisującą mapy genetyczne jest % rekombinacji wyrażany w centymorganach (cM) (1 crossing‑overcrossing‑over na 100 mejoz).
Mapa cytogenetyczna – pasmowy układ prążków charakteryzujących poszczególne chromosomy. Przedstawiana jest jako układ ciemnych i jasnych prążków, w obrębie których znajdują się sekwencje genów.
Mapa fizyczna – powstaje poprzez wykorzystanie technik biologii molekularnej do dokładnego ustalenia sekwencji badanego DNA w genomie. Jednostką opisującą mapy fizyczne są pary zasad (pz).
Konstruowanie mapy genomu

Doskonalenie technik biologii molekularnej pozwoliło na dokładne określenie lokalizacji genów i sekwencji niekodujących w genomie. Ogromny wpływ na powstanie mapy genomu człowieka miało udoskonalenie technik sekwencjonowania. Na początku rozwoju technologii badań molekularnych powszechnie wykorzystywano przede wszystkim dwie metody:
metody genetyczne – na podstawie częstości rekombinacji zachodzących między genami (w procesie crossing‑over); częstość występowania rekombinantówrekombinantów jest odwrotnie proporcjonalna do odległości między genami (więcej informacji o przebiegu i znaczeniu crossing‑over znajduje się w materiale: Częstość crossing‑over i mapowanie genówCzęstość crossing‑over i mapowanie genów); mapy genetyczne sporządzano także na podstawie analizy rodowodów, kiedy identyfikowano dziedziczność pewnych zmian z pokolenia na pokolenie w obrębie jednej rodziny.
metody fizyczne – np. analiza aberracji chromosomówaberracji chromosomów (przede wszystkim delecji, widocznych mikroskopowo) lub przez analizę efektów działania genów zmienionych mutacją.
Podczas tworzenia mapy genomu wykorzystuje się markery genetyczne. Markery to dane właściwości organizmów, pozwalające na określenie ich genotypów. Pierwotnie były to markery oparte na genach - odcinkach DNA kodujących informację o danym białku. Brak lub obecność danego genu mogła wpływać na wygląd lub funkcjonowanie organizmu. Dzięki temu możliwe było wykrycie różnic. Geny jako markery nie są jednak nadal wykorzystywane, ze względu na duże fragmenty odcinków niekodujących między nimi. Konieczne było opracowanie innych metod, opierających się na markerach molekularnych, które wykorzystują właściwości kwasów nukleinowych, głównie DNA.
Obecnie jednym z wykorzystywanych markerów w trakcie tworzenia map genetycznych jest polimorfizm długości fragmentów restrykcyjnych (RFLP, ang. restriction fragment length polymorphism). RFLP opiera się na analizie powielonego w reakcji PCR fragmentu DNA, poddanego trawieniu restrykcyjnemu. Dzięki wysokiej specyficzności używanych enzymów restrykcyjnych możliwa jest analiza wielu próbek. PolimorfizmPolimorfizm fragmentów pojawia się wtedy, gdy miejsce rozpoznawane przez enzym ulegnie zmianie. Skutkuje to brakiem cięcia sekwencji DNA i zmienionym układem analizowanych poprzez elektroforezę prążków. Więcej na ten temat znajdziesz w materiale: Analiza restrykcyjna i elektroforeza DNAAnaliza restrykcyjna i elektroforeza DNA.
Rozwój technik mapowania genomu objął także metody fizyczne. Nowe metody biologii molekularnej pozwoliły na identyfikację i ustalenie położenia genów w chromosomie za pomocą sond molekularnychsond molekularnych z wykorzystaniem zjawiska hybrydyzacji DNAhybrydyzacji DNA. Więcej informacji o działaniu sond molekularnych znajduje się w materiale: Sondy molekularne i hybrydyzacja DNASondy molekularne i hybrydyzacja DNA.
Jedną z najczęściej stosowanych technik, służących do konstrukcji map fizycznych, jest metoda fluorescencyjnej hybrydyzacji in situ (FISH, ang. fluorescence in situ hybrydization). W metodzie FISH wykorzystywana jest sonda molekularna wyznakowana fluorescencyjnie, która łączy się z sekwencją DNA. Obserwacja pod mikroskopem fluorescencyjnym pozwala na wykrycie sygnału z sondy. Umożliwia to lokalizację badanego fragmentu w określonym miejscu chromosomu.
Opisane wyżej metody pozwalają na identyfikację i lokalizację danego genu. Kolejność ułożenia genów w chromosomie ustalona tymi sposobami jest zwykle zgodna. Istnieją jednak rozbieżności między wynikami uzyskanymi metodą fizyczną i genetyczną. W wyniku sekwencjonowaniasekwencjonowania DNA udało się ustalić dokładną sekwencję genomu człowieka, obejmującą zarówno części kodujące (geny), jak i sekwencje niekodujące. Mapy genomów przyczyniły się do prawidłowego złożenia danych, które uzyskano w wyniku sekwencjonowania. W trakcie sekwencjonowania genomu człowieka ustalono kolejność nukleotydów we fragmentach analizowanego DNA. Te jednak musiały zostać ułożone w odpowiedniej kolejności, tworząc pełną sekwencję. Bez szczegółowej wiedzy na temat mapy genomu, to zadanie byłoby znacznie utrudnione. Ze względu na wykorzystane techniki, mapa genomu może wyglądać w różny sposób.
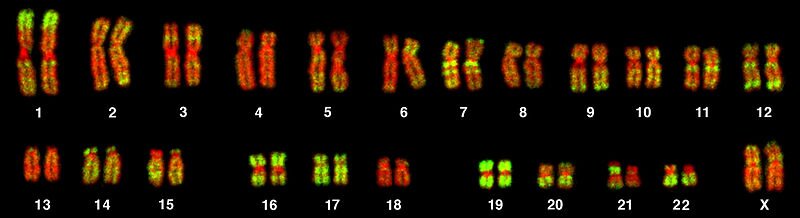
Szczegółowych informacji o lokalizacji i sekwencjach genów dostarczyło sekwencjonowanie genomu. Przełomem w badaniach ludzkiego genomu były odkrycia programu Human Genome Project.
Human Genome Project (HGP) – Projekt poznania genomu ludzkiego

W 1988 r. założono międzynarodową organizację Human Genome Organisation (HUGO), koordynującą współpracę wielu jednostek naukowych zajmujących się genomiką. Efektem prac naukowców było zapoczątkowanie Human Genome Project (HGP) – programu naukowego, który miał na celu poznanie sekwencji DNA ludzkiego genomu i identyfikację występujących w nim genów. Dawcami DNA do projektu były anonimowe osoby pochodzące z Europy, Afryki, Azji oraz Ameryki Północnej, Środkowej i Południowej. Planowano, że realizacja projektu zajmie 15 lat. Do badań genomu ludzkiego przystąpiła także firma Celera Genomics, w której zastosowano nową metodę polegającą na sekwencjonowaniu losowych fragmentów DNA. W 2000 r. ogłoszono, że poznano wstępny opis genomu człowieka, a dane uzyskane przez HGP i Celera Genomics opublikowano w dwóch niezależnych artykułach w czasopismach „Science” oraz „Nature”. W 2003 r. HGP i Celera opublikowały wspólny dokument stwierdzający zakończenie sekwencjonowania 99% genomu z trafnością 99,99%. Dane pochodzące z HGP są publicznie dostępne. Szacowany koszt badań to ok. 3 miliardy dolarów. Założyciel firmy Celera Genomics Craig Venter przyznał, że w projekcie wykorzystano próbkę jego DNA.
Poznanie genomu człowieka pozwoliło na oszacowanie liczby występujących w nim genów na ok. 22,3 tys. (początkowo zakładano istnienie ok. 100 tys. genów). Nadal są prowadzone badania nad identyfikacją i funkcją genów, a także publikowane analizy sekwencji poszczególnych chromosomów. Więcej informacji o wynikach programu Human Genome Project znajduje się w sekcji „Audiobook”.
Dodatkowym efektem pracy przy poznaniu sekwencji genomu ludzkiego było także poznanie sekwencji genów innych organizmów, w tym E. coli czy myszy. Dane te pozwoliły na analizę porównawczą i wyznaczenie stopnia podobieństwa między organizmami, a także ustalenie niezmiennych ewolucyjnie regionów DNA, które mogą posłużyć za narzędzie ustalania pokrewieństwa. Analiza genomu człowieka przyczyniła się także do rozwoju technik biologii molekularnej, w tym technik mapowania. Ważną kwestią okazał się także wynik badań na społeczeństwo i sprawy etyczne związane z poznaniem genomu człowieka. Publikacja wyników programu wywołała dyskusję m.in. na temat zagrożeń związanych z ingerencją w genom człowieka.

Słownik
duże zmiany materiału genetycznego, pociągające za sobą zmiany w liczbie lub wyglądzie chromosomów, wykrywalne w badaniach cytogenetycznych; mogą dotyczyć zarówno liczby, jak i rearanżacji strukturalnych w określonych chromosomach; aberracje chromosomów są przyczyną wielu chorób genetycznych
proces zachodzący podczas profazy mejozy I, polegający na wymianie odcinków chromatyd niesiostrzanych (zawierających tę samą informację, ale odmienną jej wersję) w miejscach określanych jako chiazmy, pomiędzy chromosomami homologicznymi
kompletny zestaw informacji genetycznej danego organizmu; u człowieka składa się z dwóch odrębnych genomów – zlokalizowanego w jądrze komórkowym genomu jądrowego oraz genomu mitochondrialnego obecnego w mitochondriach
spontaniczne łączenie się jednoniciowych kwasów nukleinowych mających komplementarną sekwencję nukleotydów
występowanie dwóch lub więcej wariantów/możliwości np. zasad azotowych w konkretnym miejscu w sekwencji DNA populacji, których częstość występowania wyklucza spontaniczną mutację
komórka lub osobnik powstały w wyniku rekombinacji genetycznej
metoda pozwalająca na określenie kolejności nukleotydów w DNA; wykorzystuje m.in. mechanizm syntezy nici z wyznakowanymi nukleotydami
wyznakowany fragment DNA lub RNA, wykorzystywany do lokalizowania komplementarnych sekwencji DNA lub RNA oraz do oceny poziomu RNA za pomocą hybrydyzacji kwasów nukleinowych