Przeczytaj
Aby prześledzić ewolucję gatunków, można przyglądać się np. skamieniałym szkieletom, jakie po nich pozostały. W ten sposób ustalono, że współczesne ptaki pochodzą od dinozaurów. Co więcej, można je zaklasyfikować do jednej z grup tych wielkich gadów i uznać za ich jedynych żyjących dziś przedstawicieli. Więcej informacji na ten temat znajdziesz w e‑materiale: Czy ptaki są dinozaurami?Czy ptaki są dinozaurami? Obecnie tworzone drzewa pokrewieństw między organizmami opierają się na analizie genomów organizmów. Rozwój technik pozwalających na badania DNA zrewolucjonizował nauki biologiczne, a systematyka organizmów uległa znacznej zmianie.
Identyfikacja materiału genetycznego
Analizy fenotypoweAnalizy fenotypowe są niedostatecznym źródłem wiedzy w przypadku ustalania pokrewieństwa. Cechy, na których opiera się analiza, mogły się wykształcić w sposób niezależny u różnych organizmów. Duży wpływ na fenotyp organizmu mają także warunki środowiska. Wiąże się z tym zjawisko plastyczności fenotypu - na podstawie danego genotypu mogą powstać różne cechy fenotypowe, w zależności od działających na organizm czynników środowiskowych. Trudne jest także ustalenie pokrewieństwa na wczesnych etapach rozwoju, ze względu na słabo wykształcone cechy specyficzne dla gatunku. Dlatego w przypadku analiz pokrewieństwa niezmiernie ważne są analizy genomów organizmów. Analizy porównawcze sekwencji genomów dostarczają wielu informacji i często podważają wyniki uzyskane na podstawie obserwacji fenotypowej. Uzyskane sekwencje są archiwizowane w bazach danych, a programy komputerowe pozwalają na ich szybkie porównywanie oraz wyszukiwanie podobieństw. Specjalistyczne narzędzia służą także do tworzenia drzew pokrewieństw oraz ustalania tempa zmian materiału genetycznego na przestrzeni lat. Problemem, który znacząco ogranicza zakres wykorzystania analiz DNA, jest brak możliwości uzyskania prób materiału genetycznego od dawno wymarłych organizmów roślin i zwierząt.

Aby móc przeanalizować pokrewieństwo organizmów na podstawie DNA oraz wyciągnąć wnioski o pokrewieństwie gatunków, należy zsekwencjonowaćzsekwencjonować materiał genetyczny. Po pozyskaniu próbki od danego organizmu należy wyizolować jego materiał genetyczny. DNA organizmu może zostać powielony w reakcji łańcuchowej polimerazy (PCR, ang. polymerase chain reaction), w wyniku czego w próbce będzie się znajdować wiele kopii DNA.
Więcej na temat PCR w e‑materiale: Łańcuchowa reakcja polimerazy.
Taki materiał, o odpowiednim stężeniu oraz czystości, można wykorzystać do sekwencjonowania. Poznaną sekwencję można porównać z analogiczną obecną u innych organizmów i na tej podstawie określić, gdzie występują różnice. Im mniej różnic, tym bardziej podobne są dwa organizmy. Naukowcy wybrali kilka genów, które szczególnie dobrze poznali, i to właśnie one są najczęściej ze sobą porównywane
Jedną z pierwszych technik pozwalających na sekwencjonowanie DNA była metoda Sangera, opierająca się na znakowanychznakowanych formach nukleotydów, które powodowały zakończenie syntezy nici. Dzięki analizie elektroforetycznejelektroforetycznej możliwy jest rozdział cząsteczek na podstawie wielkości fragmentu DNA. Podczas elektroforezy najkrótsze fragmenty migrują najszybciej, a wyznakowanie nukleotydów pozwala na określenie, który nukleotyd kończy nić. Odczyt sekwencji polegał na prześledzeniu rozdziału prążków. Obecnie cały proces został zautomatyzowany, co pozwala na analizę większych fragmentów DNA w krótszym czasie. Szczegółowe informacje na temat elektroforezy DNA znajdują się w materiale: Sekwencjonowanie DNA.
Nowoczesne metody sekwencjonowania DNA
Znakowanie fluorescencyjne nukleotydów hamujących wydłużanie DNA
Dla rozwoju metod sekwencjonowania DNA niezmiernie ważne było opracowanie techniki w oparciu o znakowanie fluorescencyjne. Pozwoliło znacznie przyspieszyć sam proces i go zautomatyzować. Ponieważ różne fluorochromyfluorochromy mają odmienne długości fal emisji po wzbudzeniu, możliwa jest obserwacja różnych kolorów fluorescencji. Ta cecha została wykorzystana w metodzie automatycznego sekwencjonowania. DideoksynukleotydyDideoksynukleotydy (ddNTP) wyznakowano bowiem różnymi fluorochromami, co pozwoliło na sekwencjonowanie z użyciem wszystkich czterech ddNTP w jednej probówce. W związku z tym wykorzystuje się jedną mieszaninę substratów reakcji, a nie jak w metodzie Sangera cztery probówki, gdzie każda miała inny rodzaj ddNTP. W technice tej wykorzystuje się detektor, mający zdolność do zbierania i odróżniania sygnałów płynących z fluorochromów.
Znakowanie fluorescencyjne opiera się na elektroforezie kapilarnej w automatycznym sekwenatorze. Wykonywana jest ona w kwarcowych kapilarach o średnicy 25–100 mikrometrów. Na ich końcu znajduje się wspomniany detektor. Sekwencja dzięki różnym sygnałom fluorochromów może być odczytywana przez komputer „na bieżąco”, co chroni przed pomyleniem kolejności prążków na żelu, zdarzającym się w tradycyjnej metodzie Sangera. Wynik takiego sekwencjonowania to chromatogramchromatogram, na którym widoczne są piki dla poszczególnych nukleotydów, wyznakowanych różnymi fluorochromami.
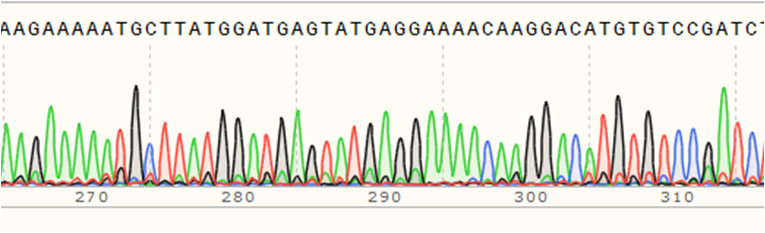
Sekwencjonowanie nowej generacji (NGS, ang. new generation sequencing)
Sekwencjonowanie NGS pozwala na sekwencjonowanie DNA pobranego np. z próbki gleby czy wody. Wiąże się to z ogromną ilością różnych sekwencji DNA, które są odczytywane w tym samym czasie. W wielu przypadkach poznajemy nieznane wcześniej nauce gatunki.
Sekwencjonowanie nie odbywa się w probówce, ale na płytce, na powierzchni której znajdują się dołki. DNA w dołku jest powielane przy użyciu reakcji PCR, tworząc zbiór fragmentów. W technice tej stosuje się wyznakowane fluorescencyjnie nukleotydy. Każdy z nukleotydów wyznakowany jest innym kolorem, co umożliwia ustalenie, jaki nukleotyd został przyłączony. Cykl taki powtarza się do 200 razy, aby otrzymać całkowitą sekwencję fragmentu. W tym samym czasie analizowana jest ogromna liczba próbek, która daje od 1 mln do 43 mld sekwencji na analizę. Wyniki odczytów są później analizowane przez programy komputerowe.
Głównymi zaletami najnowszych metod sekwencjonowania są:
wysoka przepustowość;
niski koszt analizy jednej próbki;
możliwość analizy ogromnej ilości danych w stosunkowo krótkim czasie (technologie NGS generują dużą liczbę wyników, które następnie należy przetwarzać, wykorzystując innowacyjne narzędzie bioinformatyczne oraz komputery charakteryzujące się dużą mocą obliczeniową);
odstąpienie od radioaktywnych izotopów i zastąpienie ich nukleotydami znakowanymi fluorescencyjnie;
skrócenie czasu całego procesu, dzięki czemu większa ilość materiału może zostać przeanalizowana i wprowadzona do ogólnodostępnych baz danych, a na ich podstawie można następnie próbować ustalić stopień pokrewieństwa badanych organizmów z już znanymi gatunkami, lub stwierdzić, iż próbka materiału genetycznego pochodzi z jeszcze nieznanego gatunku.
Ustalanie pokrewieństwa
Poznany za pomocą sekwencjonowania ciąg nukleotydów w badanym DNA jest wprowadzany do bazy danych. Współcześnie istnieje wiele takich baz. Gromadzą one informacje o genomach wielu gatunków lub wybranych grup organizmów, np. drożdży, bakterii czy roślin. Bazy danych pozwalają nie tylko na sprawdzenie, czy w genomie danego organizmu występuje dana sekwencja, ale także na poznanie białka, które jest przez nią kodowane.
Ustalanie pokrewieństwa między organizmami opiera się na analizie porównawczej sekwencji genomów organizmów. W toku ewolucji w DNA organizmów pojawiają się mutacje, ale w jednych jego fragmentach występują częściej, a w innych rzadziej. Sekwencje, w których rzadko dochodzi do mutacji, określane są mianem konserwatywnych i to ich używa się najczęściej do tego rodzaju porównań. Zazwyczaj sekwencje konserwatywne kodują bardzo ważne dla przeżycia komórki białka. Do analiz porównawczych wykorzystywane są także sekwencje aminokwasowe białek. Część zmian w genach nie skutkuje bowiem zmianą kodowanych przez nie aminokwasów, przez co białka utrzymują wysoki stopień podobieństwa.
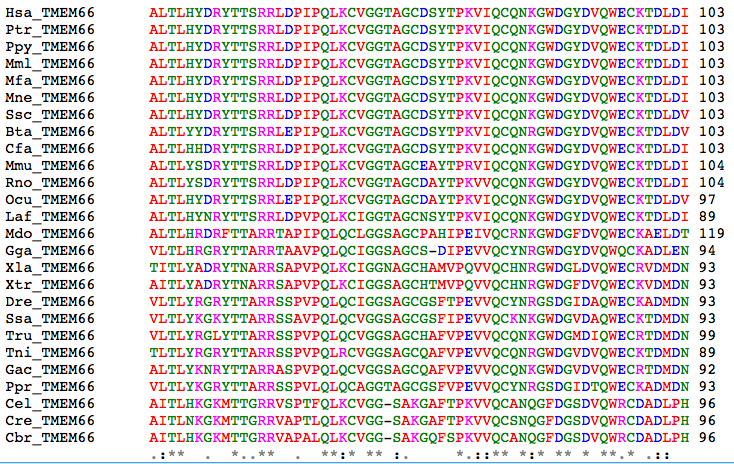
Wyniki analizy porównawczej pozwalają na określenie stopnia podobieństwa porównywanych sekwencji, a na tej podstawie można wykonać drzewo filogenetycznedrzewo filogenetyczne. Im większy stopień homologiihomologii, tym bliżej spokrewnione powinny być organizmy. W celu dokładnego określenia stopnia pokrewieństwa należy porównać długie sekwencje. Porównanie krótkich fragmentów może dać błędny wynik.
Analiza pokrewieństwa w obrębie taksonów (blisko spokrewnionych organizmów) może się opierać na krótkich fragmentach. W takim przypadku wykorzystywany jest tzw. barkoding. Więcej o metodzie barkodingu w materiale: Barkoding DNA - analiza tekstu naukowegoBarkoding DNA - analiza tekstu naukowego.
Konstruowanie drzew filogenetycznych
Do tworzenia drzew filogenetycznych wykorzystywane są programy komputerowe oraz narzędzia dostępne bezpośrednio w bazach danych, gromadzących informacje o sekwencjach DNA. Jedną z najczęściej używanych baz danych jest NCBI (ang. National Center for Biotechnology Information). Na stronie internetowej NCBI znajduje się specjalna zakładka poświęcona taksonomii oraz ustalaniu homologii. Do analizy można użyć danych zawartych w bazie, jak również własnych wyników. Drzewa filogenetyczne wykonane w różnych programach mogą się jednak różnić, ze względu na różne modele wykorzystywane do analiz. Więcej na ten temat znajdziesz w e‑materiale: Kontruowanie drzew filogenetycznychKontruowanie drzew filogenetycznych.
Do analizy pokrewieństwa często wykorzystywany jest rRNA - rybosomalny RNA, który wchodzi w skład podjednostek rybosomalnych. Szczególnie często stosowany jest fragment 16S podjednostki rybosomalnej, który jest wysoce konserwatywny. Jest to jedynie jedna z wielu cząsteczek, służących ustalaniu pokrewieństwa. Różne grupy organizmów mogą wykazywać większą zmienność w obrębie innych cząsteczek. Przykładem porównywanej sekwencji konserwatywnej jest mitochondrialny gen kodujący cytochrom ccytochrom c, który mają rośliny, zwierzęta i wiele mikroorganizmów. U blisko spokrewnionych organizmów jest on bardzo podobny lub nawet identyczny, np. u ludzi i szympansów. Między dalszymi krewnymi różni się proporcjonalnie do stopnia pokrewieństwa. Jeżeli w wyizolowanym materiale genetycznym znajdzie się ten właśnie gen, to można z bardzo dużym prawdopodobieństwem ustalić taksonomiczną pozycję badanego organizmu.
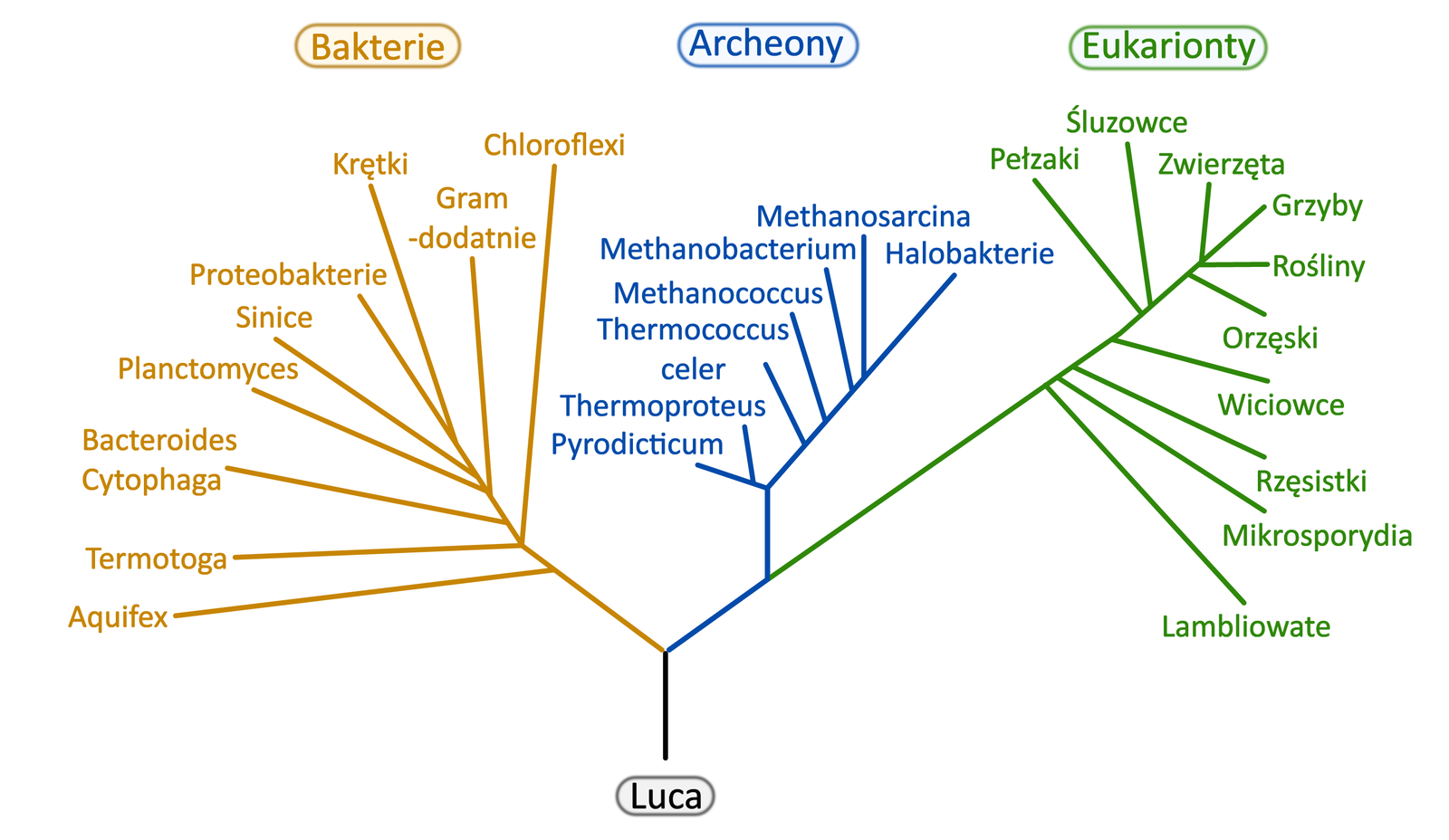
Początków drzewa filogenetycznego upatruje się w hipotetycznym organizmie nazwanym z jęz. ang. last universal common ancestor (LUCA), co oznacza 'ostatni uniwersalny wspólny przodek'. Nie był to pierwszy żywy organizm na Ziemi, ale ostatni, od którego pochodzą wszystkie pozostałe. Jeśli wyobrazimy sobie pierwsze organizmy jako pień drzewa życia, LUCA będzie ostatnim z nich przed pierwszym rozgałęzieniem pnia. Zdaniem naukowców był to jednokomórkowiec podobny do dzisiejszych archeonów.
Słownik
badania biorące pod uwagę jedynie widoczne cechy organizmu, jak liczba kończyn, kształt kości czy kolor sierści
graficzne przedstawienie procesu rozdziału cząsteczek
białko obecne u wielu organizmów, będące jednym z przenośników tlenu w łańcuchu oddechowym; wykorzystywane jest w badaniach filogenetycznych do określania stopnia pokrewieństwa między organizmami
nukleotyd hamujący wydłużanie nici DNA; nie posiada grupy -OH w pozycji 3' pentozy
graficzne przedstawienie filogenezy – ewolucyjnych zależności, stosunków pokrewieństwa między różnymi grupami systematycznymi (taksonami)
rozdział białek lub łańcuchów DNA i RNA w specjalnym żelu; umieszcza się go wraz z próbkami w polu elektrycznym, pod którego wpływem przesuwają się naprzód i rozdzielają pod względem wielkości
cząsteczka posiadająca zdolność do fluorescencji; w wyniku wzbudzenia promieniowaniem o danej długości fali emituje falę o innej długości
(gr. homólogos – zgodny) podobieństwo genów i białek wynikające z odziedziczenia po wspólnym przodku
metoda pozwalająca na określenie kolejności nukleotydów w DNA; wykorzystuje m.in. mechanizm syntezy nici z wyznakowanymi nukleotydami
nukleotyd, do którego dołączony został związek barwny lub izotop radioaktywny (inny do każdego nukleotydu) w taki sposób, aby można go potem było zidentyfikować w gotowej nici DNA