Choroby genetyczne człowieka
Jednogenowe choroby człowieka
Wymienisz jednogenowe choroby człowieka.
Określisz na podstawie analizy rodowodu podłoże genetyczne chorób człowieka: mukowiscydoza, fenyloketonuria, pląsawica Huntingtona, hemofilia, daltonizm.
Geny to instrukcja obsługi organizmu, która w dużej mierze determinuje jego cechy takie jak np. grupa krwi czy kolor oczu. Jednak ogólne zdrowie i równowaga wewnętrzna organizmu zależą od stałej współpracy genów ze środowiskiem. Jeśli w zapisie genetycznym pojawi się błąd, czyli mutacja, może dojść do rozwoju choroby genetycznej. W zależności od zakresu mutacji wyróżnia się choroby:
jednogenowe – wywołane mutacjami w obrębie jednego genu;
wielogenowe – wywołane mutacjami w obrębie różnych genów i indukowane czynnikami środowiska;
chromosomalne – wywołane mutacjami w strukturze lub liczbie chromosomów.
Znajomość prawidłowej struktury genów i chromosomów pozwala diagnozować choroby genetyczne, a rozwój metod inżynierii genetycznej otwiera drogę do ich leczenia bezpośrednio na poziomie DNA.
Choroby jednogenowe
Choroby jednogenowe są w większości spowodowane mutacjami genów zlokalizowanych w jądrowym DNA. Mogą one być dziedziczone autosomalnie lub w sprzężeniu z płcią oraz recesywnie lub dominująco.
Choroby jednogenowe dziedziczone autosomalnie recesywnie
Choroby dziedziczone autosomalnie recesywnie najczęściej dotyczą genów kodujących enzymy. Brak lub niedobór prawidłowej formy enzymu skutkuje zaburzeniami przemian metabolicznych organizmu.
Do wystąpienia objawów choroby konieczna jest obecność dwóch recesywnych alleli (a) danego genu. Choroba ujawnia się u homozygot recesywnych (aa), natomiast homozygoty dominujące (AA) i heterozygoty (Aa) nie wykazują objawów choroby. Heterozygoty są jednak nosicielami wadliwego genu, który mogą przekazać potomstwu. Choroby jednogenowe dziedziczone autosomalnie recesywnie występują z jednakową częstością u obu płci.
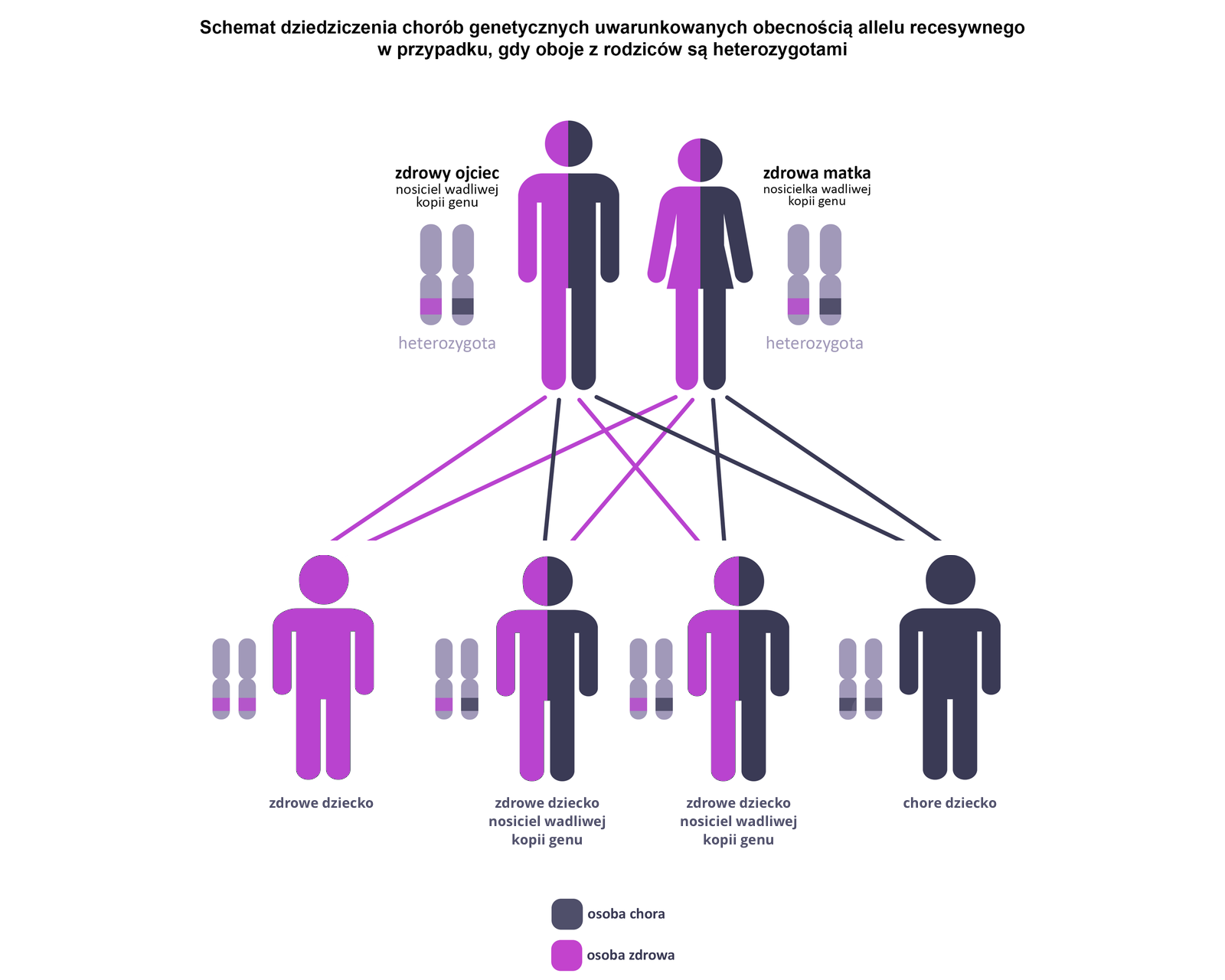
Niektóre choroby są związane z mutacjami w genach kodujących białka enzymatyczne w szlakach metabolicznych. Zablokowanie tych szlaków – spowodowane brakiem lub nieprawidłową strukturą enzymów katalizujących poszczególne reakcje nazywane jest blokiem metabolicznym. Ich skutkiem są wrodzone wady metabolizmu o podłożu genetycznym, najczęściej dziedziczone autosomalnie recesywnie. Ujawniają się w krótkim czasie po urodzeniu. Bloki metaboliczne mogą dotyczyć metabolizmu białek, tłuszczów bądź węglowodanów.
Główny szlak metabolizmu fenyloalaniny obejmuje szereg reakcji enzymatycznych prowadzących do jej przekształcenia w inne związki, m.in. tyrozynę, które są niezbędne do prawidłowego funkcjonowania organizmu. Zaburzenia tego szlaku (blok metaboliczny) mogą prowadzić do poważnych chorób, takich jak fenyloketonuria, alkaptonuria i albinizm.
Fenyloketonuria
Fenyloketonuria to choroba związana z zaburzeniami przemian metabolicznych aminokwasu fenyloalaninyfenyloalaniny. Najczęstszą przyczyną choroby są mutacje genu PAH kodującego białko – hydroksylazę fenyloalaniny (PAH). Białko to jest enzymem katalizującym przekształcenie fenyloalaniny do innego aminokwasu - tyrozynytyrozyny. Skutkiem mutacji jest niedobór lub brak białka PAH, czego konsekwencją jest gromadzenie się we krwi i płynach ustrojowych nadmiaru fenyloalaniny i produktów alternatywnego przekształcania tego aminokwasu (np. kwas fenylopirogronowy).
Nadmiar fenyloalaniny ma toksyczny wpływ na rozwój ośrodkowego układu nerwowego. Choroba objawia się już we wczesnym niemowlęctwie, wymiotami oraz nieprzyjemnym zapachem moczu i potu. W późniejszym okresie obserwuje się opóźnienie rozwoju psychoruchowego, małogłowie, niepełnosprawność intelektualną, zaburzenia neurologiczne.

W trzeciej dobie życia u noworodków wykonuje się test przesiewowy sprawdzający poziom fenyloalaniny we krwi. W przypadku potwierdzenia fenyloketonurii wprowadza się leczenie dietetyczne. Restrykcyjna dieta niskofenyloalaninowa ma na celu utrzymanie odpowiedniego poziomu tego aminokwasu we krwi i płynach ustrojowych. Wczesne rozpoznanie choroby i wdrożenie odpowiedniej diety zapobiega uszkodzeniom ośrodkowego układu nerwowego i zapewnia prawidłowy rozwój psychofizyczny dziecka.

Albinizm
Albinizm to choroba związana z zaburzeniami w procesie syntezy melanin. Przyczyną albinizmu są mutacje genu TYR kodującego tyrozynazę – enzym katalizujący reakcję przekształcania tyrozyny do 3,4‑dihydroksyfenyloalaniny, związku wykorzystywanego do syntezy melanin.

Skutkiem mutacji jest niedobór lub brak barwnika w skórze, włosach i tęczówce oka. Choroba objawia się bardzo jasną skórą, białymi włosami i różowymi lub niebieskimi tęczówkami oczu. Dodatkowo u wielu albinotycznych osobników występuje oczopląs, światłowstręt i zmniejszona ostrość wzroku. Ludzie cierpiący na albinizm wykazują nadmierną wrażliwość na promieniowanie słoneczne, co zwiększa ryzyko poparzeń słonecznych i nowotworów skóry.

Alkaptonuria
Alkaptonuria to choroba związana z zaburzeniami przemian metabolicznych kwasu homogentyzynowego. Najczęstszą przyczyną choroby są mutacje genu HGD kodującego enzym – oksygenazę homogentyzynianową (HGD) katalizujący przekształcenie kwasu homogentyzynowego do produktów cyklu Krebsa.
Skutkiem mutacji jest gromadzenie się we krwi i płynach ustrojowych nadmiaru kwasu homogentyzynowego. Jego nadmiar ma szkodliwy wpływ na tkankę chrzęstną.
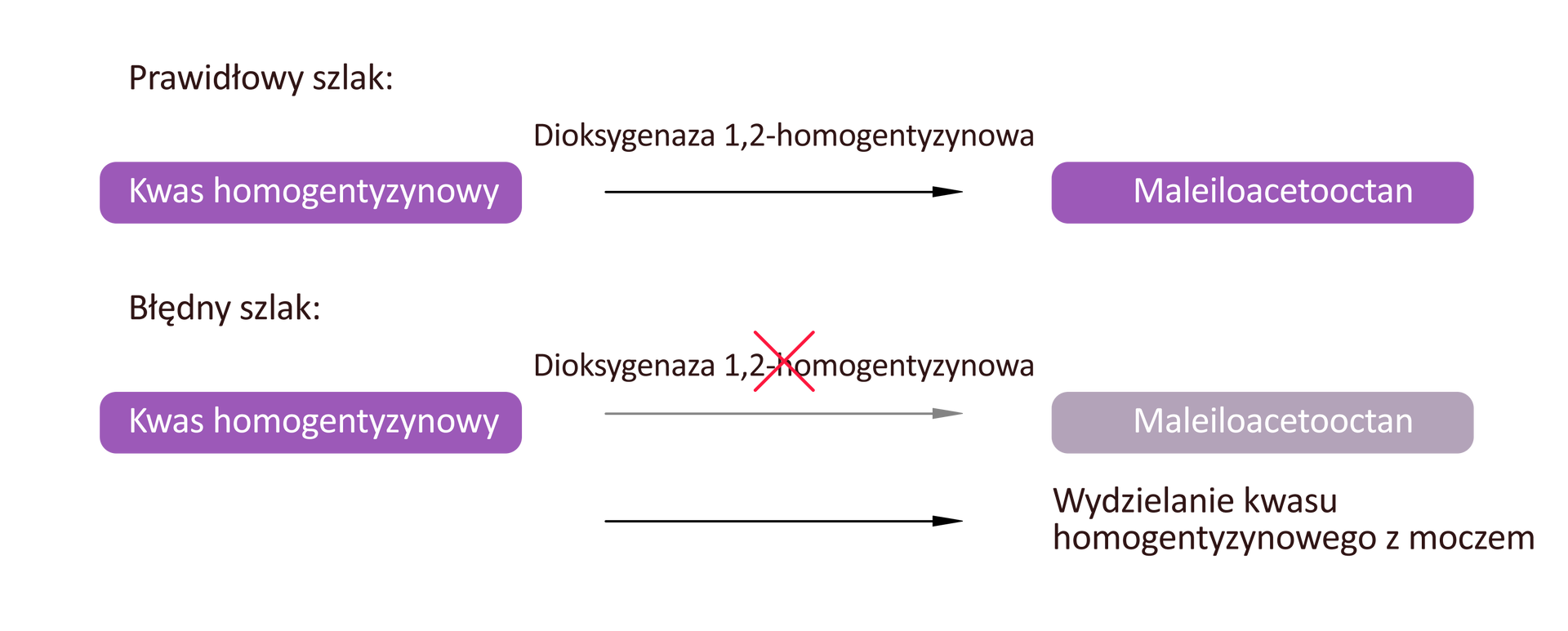
Choroba objawia się we wczesnym dzieciństwie niebiesko zabarwionym moczem spowodowanym dużym stężeniem kwasu homogentyzynowego, który ciemnieje po zetknięciu z powietrzem. W późniejszym czasie dochodzi do rozwoju stanów zapalnych i zmian zwyrodnieniowych stawów, co przejawia się bólem i sztywnością stawów oraz trudnościami w poruszaniu się.
Do niebezpiecznych chorób jednogenowych dziedziczonych recesywnie należą również mukowiscydoza, anemia sierpowata i galaktozemie.
Mukowiscydoza to choroba związana z zaburzeniem transportu jonów chlorkowych przez błony komórkowe. Przyczyną choroby są mutacje genu CFTR kodującego białko – przezbłonowy regulator transportu jonów (CFTR)przezbłonowy regulator transportu jonów (CFTR). Białko CFTR występuje na powierzchni komórek tkanki nabłonkowej dróg oddechowych, układu pokarmowego, trzustki, wątroby i gruczołów potowych. Skutkiem mutacji jest brak białka CFTR lub zaburzenie jego funkcji. W rezultacie jony chlorkowe nie są transportowane na zewnątrz komórek nabłonka, natomiast jony sodowe oraz woda są pobierane w nadmiarze do ich wnętrza. Prowadzi to do zmniejszenia zawartości wody w śluzie na powierzchni nabłonka, który staje się gęsty i lepki.
Gęsty i lepki śluz zalega w drogach oddechowych, sprzyja rozwojowi chorobotwórczych mikroorganizmów, prowadząc do częstych i trudnych w leczeniu infekcji. Konsekwencją tego jest niewydolność oddechowa. Z kolei śluz gromadzący się w przewodach wyprowadzających trzustki utrudnia odpływ soku trzustkowego, przyczyniając się do rozwoju stanów zapalnych narządu i jego zwłóknienia. Nieprawidłowe funkcjonowanie nabłonka jelit oraz upośledzenie funkcji trzustki powodują zaburzenia wchłaniania substancji pokarmowych. W przypadku gruczołów potowych obserwuje się nadmierne wydzielanie wraz z potem jonów chlorkowych i sodowych. U mężczyzn choroba prowadzi do niedrożności lub zaniku nasieniowodów i w konsekwencji do bezpłodności.
Leczenie mukowiscydozy polega na regularnym odsysaniu śluzu z dróg oddechowych, przyjmowaniu leków przeciwzapalnych oraz ułatwiających trawienie.

Anemia sierpowata to choroba związana z zaburzeniem syntezy hemoglobiny. Przyczyną choroby jest mutacja genu HBB kodującego łańcuch beta hemoglobiny. Mutacja punktowa zamiany jednego nukleotydu (tyminowego na adeninowy) w genie kodującym beta hemoglobinę skutkuje zmianą jednego aminokwasu – kwasu glutaminowego – na walinę, czego konsekwencją jest powstanie hemoglobiny S. Nieprawidłowo zbudowana hemoglobina S agreguje w erytrocytach, powodując zmianę ich kształtu na sierpowaty.
Hemoglobina S ma mniejsze powinowactwo do tlenu niż prawidłowa hemoglobina A. Dodatkowo erytrocyty sierpowate mają krótszy czas życia, w porównaniu z krwinkami prawidłowymi. Dlatego też, choroba objawia się niedokrwistością i niedotleniem tkanek organizmu.
Leczenie anemii sierpowatej polega na łagodzeniu objawów niedotlenienia.

Galaktozemia to choroba związana z zaburzeniami przemian metabolicznych węglowodanów. Najczęstszą przyczyną choroby są mutacje genu GALT kodującego białko – urydylotransferazę galaktozo‑1-fosforanu (GALT).
Białko GALT jest enzymem katalizującym przekształcenie galaktozy do glukozy. Skutkiem mutacji jest niedobór lub brak białka GALT, czego konsekwencją jest gromadzenie w tkankach ciała galaktozy i jej pochodnych. Związki te wykazują toksyczny wpływ na organizm, w szczególności na tkankę nerwową.
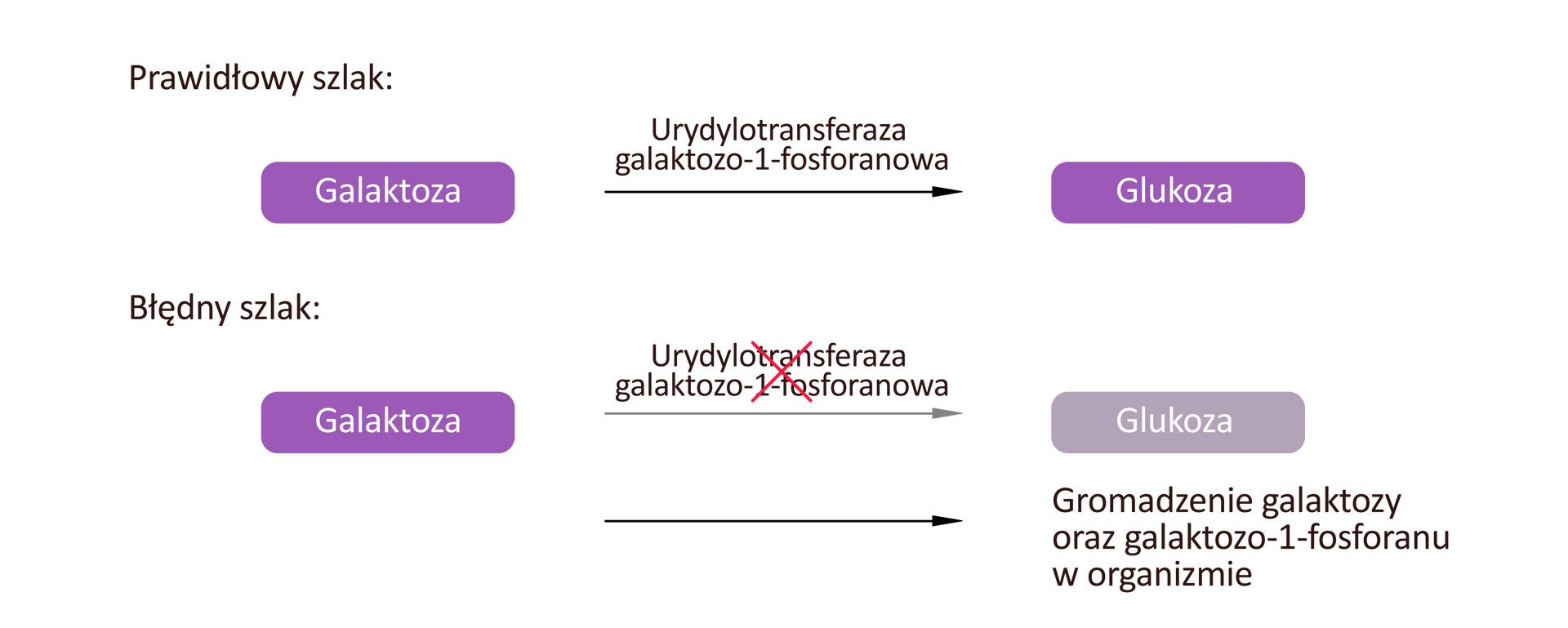
Choroba objawia się tuż po narodzinach niechęcią noworodka do karmienia, wymiotami, biegunką. W późniejszym czasie pojawiają się senność, apatia, brak przyrostu masy ciała, powiększenie wątroby i śledziony, upośledzenie pracy nerek, zaburzenia neurologiczne, opóźnienie rozwoju umysłowego.
Wczesne rozpoznanie choroby i zastosowanie restrykcyjnej diety wykluczającej spożycie mleka i jego przetworów pozwalają złagodzić objawy choroby.
Choroby jednogenowe dziedziczone autosomalnie dominująco
Choroby dziedziczone autosomalnie dominująco występują w ludzkiej populacji znacznie rzadziej niż choroby dziedziczone autosomalnie recesywnie. Do wystąpienia objawów choroby wystarcza tylko jeden wadliwy allel (A) danego genu. Choroba ujawnia się u heterozygot (Aa) i homozygot dominujących (AA), przy czym homozygoty dominujące często wykazują cięższy przebieg choroby lub mutacja jest dla nich letalnaletalna. Choroby jednogenowe dziedziczone autosomalnie dominująco występują z jednakową częstością u obu płci.

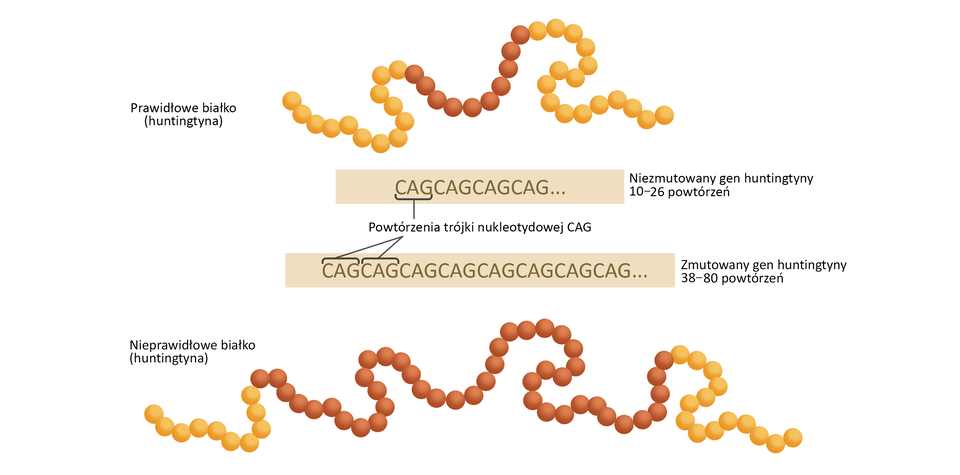
Choroba Huntingtona jest związana z zaburzeniami pracy komórek nerwowych. Przyczyną choroby są mutacje genu IT‑15 kodującego huntingtynęhuntingtynę. Białko to występuje m.in. w komórkach nerwowych i uczestniczy w przekazywaniu impulsów nerwowych oraz reguluje przebieg procesów transkrypcji, translacji i transportu komórkowego.
Skutkiem mutacji jest obecność białka o nieprawidłowej strukturze, które tworzy złogi w neuronach. Odkładanie się nieprawidłowego białka powoduje obumieranie komórek nerwowych. Choroba objawia się około 30–40 roku życia, czemu towarzyszą: spadek masy ciała, zaburzenia psychiczne, zmiany nastroju, trudności w pisaniu i uczeniu się, zaburzenia pamięci. W późniejszym czasie pojawiają się niekontrolowane ruchy twarzy, kończyn i tułowia.
Choroby sprzężone z płcią dziedziczone recesywnie
Choroby dziedziczone recesywnie sprzężone z płcią, dotyczą genów położonych na chromosomie X. Choroby te, znacznie częściej występują u mężczyzn niż u kobiet, co wynika z obecności u nich tylko jednej kopii genu sprzężonego z chromosomem X. Kobiety chorują rzadziej, gdyż mają dwie kopie genu sprzężonego z chromosomem X. Heterozygotyczne kobiety są nosicielami wadliwego genu, który mogą przekazać potomstwu, natomiast same nie wykazują objawów choroby. Kobiety chorują w homozygotycznym układzie genów recesywnych, ale taka możliwość zachodzi bardzo rzadko.

Hemofilia, zwana także krwawiączką, to choroba polegająca na zaburzeniu krzepnięcia krwi, związana z niedoborem czynników krzepnięcia (VIII lub IX). Jej konsekwencją jest niezdolność organizmu do naturalnego zatrzymywania krwawień.
Objawy pojawiają się we wczesnym dzieciństwie. Należą do nich: skłonność do siniaków, przedłużające się krwawienia po zabiegach, samoistne krwawienia do stawów i mózgu. Wylewy krwi do stawów wywołują ból i powodują uszkodzenie błony maziowej, czego skutkiem jest ograniczenie ruchomości stawu. Wylewy do mózgu mogą skutkować długotrwałymi bólami głowy, drgawkami lub obniżonym poziomem świadomości.
Leczenie hemofilii polega na powtarzalnym podawaniu brakującego czynnika krzepnięcia krwi w formie dożylnej. Wyniki testów pokazują, że prawdopodobna jest terapia genowa hemofilii.
W ostatnich latach zatwierdzono pierwsze terapie genowe dla niektórych rodzajów hemofilii. Polegają one na wprowadzeniu do komórek wątroby pacjenta poprawnej kopii genu brakującego czynnika krzepnięcia, dzięki czemu organizm sam zaczyna go produkować.


Daltonizm to zespół różnych jednostek chorobowych związanych z zaburzeniami widzenia barwy czerwonej i zielonej, uwarunkowanych nieprawidłowościami w obrębie dwóch genów leżących obok siebie na długim ramieniu chromosomu X. Przyczyną szeroko rozumianego daltonizmu są częściowe lub całkowite delecje w obrębie genów kodujących barwniki wzrokowe.
W populacji europejskiej zaburzenia widzenia barwy czerwonej i zielonej występują u około 8% mężczyzn i 0,5% kobiet. U osób rasy czarnej i żółtej zaburzenia postrzegania kolorów występują znacznie rzadziej.



Dystrofia mięśniowa Duchenne'a jest jednym z najczęstszych i najcięższych w przebiegu typów dystrofii mięśniowej – występuje z częstością 1:3600 urodzonych mężczyzn. Przyczyną choroby są mutacje w genie białka dystrofiny zlokalizowanego na krótkim ramieniu chromosomu X (gen ten jest największym zidentyfikowanym genem w ludzkim genomie).
Skutkiem mutacji jest brak lub niedobór dystrofiny. Białko to występuje w miocytach mięśni szkieletowych, serca i gładkich, oraz w komórkach nerwowych. Zasadniczą rolą dystrofiny jest stabilizacja błony komórkowej komórek mięśniowych i nerwowych. Brak lub niedobór dystrofiny prowadzi do martwicy miocytów oraz zaburzeń w funkcjonowaniu układu nerwowego.
Objawy choroby w postaci osłabienia mięśni pojawiają się zwykle w 3–4 roku życia i szybko się pogłębiają. Zanik mięśni rozpoczyna się w udach i miednicy, następnie obejmuje ramiona. Początkowo może to powodować problemy ze wstawaniem, ale większość osób dotkniętych tą chorobą nie chodzi samodzielnie już w wieku ok. 12 lat. W tym wieku dochodzi także do osłabienia mięśnia sercowego i mięśni oddechowych, co nasila problemy krążeniowo‑oddechowe. Dystrofię mięśniową Duchenne’a można wykryć od razu po urodzeniu za pomocą testów genetycznych, ale także poprzez określenie poziomu tzw. kinazy kreatynowej we krwi i poziomu kreatyny w moczu.
Choroby sprzężone z płcią dziedziczone dominująco
Choroby dziedziczone dominująco sprzężone z chromosomem X występują znacznie rzadziej w ludzkiej populacji niż choroby recesywne sprzężone z płcią. Należy do nich m.in. krzywica oporna na witaminę DIndeks dolny 33. W tym typie dziedziczenia choroba u mężczyzn przebiega z ciężkimi objawami i zazwyczaj jest letalna. U heterozygotycznych kobiet obraz kliniczny choroby jest zmienny i uzależniony od losowej inaktywacji chromosomu Xlosowej inaktywacji chromosomu X w komórkach ciała.
Krzywica oporna na witaminę DIndeks dolny 33 to choroba związana z zaburzeniami gospodarki wapniowo‑fosforanowej, pojawiająca się z częstością 1:20 000 urodzeń. Konsekwencją niedoboru fosforanów jest słaba mineralizacja tkanki kostnej, deformacje kości i zaburzenia wzrostu kośćca.

Przyczyną choroby jest mutacja genu PHEX zlokalizowanego na chromosomie X. Gen PHEX ulega ekspresji w komórkach kości i zębów, a powstające białko enzymatyczne wpływa na regulację poziomu fosforanów. Brak funkcjonalnego białka zwiększa wydalanie fosforanów wraz z moczem oraz zmniejsza syntezę witaminy DIndeks dolny 33.
Objawy choroby pojawiają się w pierwszych dwóch latach życia i dotyczą zniekształceń kończyn dolnych, zaburzeń proporcji ciała, zahamowania wzrostu.
W dalszym przebiegu choroby pojawiają się deformacje klatki piersiowej i kręgosłupa. Leczenie polega na doustnym podawaniu fosforanów i suplementacji witaminy DIndeks dolny 33.
Podsumowanie
Choroby genetyczne to schorzenia spowodowane mutacjami w materiale genetycznym (DNA). W zależności od rodzaju i lokalizacji mutacji wyróżnia się:
- choroby jednogenowe – wynikające z mutacji w jednym genie,
- wielogenowe – wywołane mutacjami w obrębie różnych genów i indukowane czynnikami środowiska;
- choroby chromosomowe – związane ze zmianą liczby lub struktury chromosomów,Choroby jednogenowe mogą dziedziczyć się jako:
- autosomalne dominujące,
- autosomalne recesywne,
- sprzężone z chromosomem X (dominujące lub recesywne).Mukowiscydoza – choroba autosomalna recesywna. Spowodowana mutacją genu CFTR, prowadzi do zaburzeń transportu jonów chlorkowych i powstawania gęstego, lepkiego śluzu w układzie oddechowym i pokarmowym. Występuje z jednakową częstością u dziewczynek i chłopców.
Fenyloketonuria – choroba autosomalna recesywna. Wynika z zaburzenia przemian metabolicznych aminokwasy fenyloalaniny, co prowadzi do jej nagromadzenia i uszkodzenia układu nerwowego. Dotyczy obu płci.
Pląsawica Huntingtona - choroba autosomalna dominująca. Jest skutkiem mutacji w genie HTT. Prowadzi do postępującego uszkodzenia układu nerwowego, zaburzeń ruchowych i psychicznych. Do ujawnienia choroby wystarczy jedna zmutowana kopia genu. Chorują zarówno kobiety, jak i mężczyźni.
Hemofilia – choroba recesywna sprzężona z chromosomem X. Powoduje niedobór czynników krzepnięcia co skutkuje zaburzeniami krzepnięcia krwi. Najczęściej chorują chłopcy, ponieważ mają tylko jeden chromosom X. Dziewczynki zwykle są nosicielkami i chorują rzadko (muszą mieć dwie zmutowane kopie genu).
Daltonizm – choroba recesywna sprzężona z chromosomem X. Polega na zaburzeniu rozpoznawania barw. Podobnie jak hemofilia, częściej występuje u chłopców niż u dziewczynek.
Ćwiczenia utrwalające
Osoby cierpiące na daltonizm: Możliwe odpowiedzi: 1. nie widzą żadnych kolorów, 2. nie rozróżniają niektórych barw, 3. to głównie mężczyźni, 4. to głównie kobiety
Heterozygotyczna kobieta, chorująca na pląsawicę Huntingtona, ma czworo dzieci ze zdrowym mężczyzną. Uzupełnij krzyżówkę genetyczną i wykonaj poniższe polecenia.
Określ sposób dziedziczenia pląsawicy Huntingtona.
Podaj prawdopodobieństwo wystąpienia pląsawicy Huntingtona u dzieci opisanej pary.
Wróć do polecenia na stronie „Na dobry początek” i dopisz brakujące definicje. Pamiętaj, żeby nie kopiować słownika, ale wyjaśnić każde słowo kluczowe w miarę możliwości swoimi słowami.