Przeczytaj
Choroby bloku metabolicznego

Bloki metaboliczne zostały opisane po raz pierwszy na początku XX w. przez Archibalda Garroda, który badał zaburzenia przemiany kwasu homogentyzynowego w rodzinie dotkniętej alkaptonurią. Wysnuł on wniosek, że choroba ta wynika z zahamowania jednego z etapów przemiany biochemicznej, co spowodowane jest brakiem aktywności określonego enzymu. Zaprezentował całość swoich prac w wykładzie pt. Wrodzone wady metabolizmu (ang. Errors of Metabolism) przed Royal College of Physicians (Królewskim Kolegium Lekarzy).
Do dzisiaj opisano setki różnych wrodzonych błędów metabolizmu, z których większość to choroby rzadkie. Zazwyczaj dziedziczą się recesywnierecesywnie autosomalnie lub w sprzężeniu z chromosomem X. Zalicza się do nich nieprawidłowości metabolizmu aminokwasów, węglowodanów, tłuszczów, kwasów organicznych oraz cyklu mocznikowego, szlaków przemian energii i transportu jonów metali.
Większość przypadków choroby występuje w wyniku dziedziczenia autosomalnego recesywnego, co oznacza, że choroba występuje u osoby, która odziedziczyła dwa wadliwe allele genu (recesywne) – od matki i od ojca. Rodzice, posiadający jeden prawidłowy a drugi wadliwy allel genu, nie chorują, a są jedynie nosicielami. Rzadziej choroba powstaje u potomstwa osób zdrowych, niebędących nosicielami w wyniku wystąpienia nowej mutacji.
Przykładowe zaburzenia przemian metabolicznych uwarunkowane genetycznie
Nazwa choroby | Częstość | Produkt zmutowanego genu | Lokalizacja chromosomowa genu |
|---|---|---|---|
Nieprawidłowości metabolizmu aminokwasów | |||
Fenyloketonuria | 1/7 tys. | brak enzymu hydroksylazy fenyloalaninowej | 12q24 |
Alkaptonuria | 1/250 tys. | brak enzymu dioksygenazy 1,2‑homogentyzynowej | 3q2 |
Albinizm | 1/35 tys. | brak enzymu tyrozynazy | 11q |
Choroba syropu klonowego | 1/180 tys. | brak kompleksu dehydrogenazy alfa‑ketokwasów o rozgałęzionym łańcuchu | wiele loci |
Nieprawidłowości metabolizmu węglowodanów | |||
Galaktozemia | 1/35 tys. do 1/60 tys. | brak enzymu urydylotransferazy galaktozo‑1‑fosforanowej | 9q13 |
Fruktozuria | 1/100 tys. | brak enzymu aldolazy fruktozo‑1‑fosforanu | 2p23 |
Najlepiej poznanymi blokami metabolicznymi u ludzi są nieprawidłowości w przemianie fenyloalaninyfenyloalaniny oraz tyrozynytyrozyny.
Fenyloketonuria
Najlepiej poznaną chorobą wywołaną zaburzeniami metabolizmu aminokwasów jest fenyloketonuria. Do jej wystąpienia przyczynia się brak lub niedobór hydroksylazy fenyloalaninowej – enzymu, który przekształca fenyloalaninę w tyrozynę. Wskutek mutacji genowej (substytucji, insercji lub delecji) w genie PAHgenie PAH dochodzi do braku lub obniżonej aktywności tego enzymu. Prowadzi to do gromadzenia się fenyloalaniny we wszystkich płynach ustrojowych i przekształcania jej do toksycznego kwasu fenylopirogronowego.

U zdrowych ludzi 3/4 fenyloalaniny jest przekształcane w tyrozynę, a pozostała 1/4 – włączana do białek. Ze względu na to, że u osób chorych na fenyloketonurię główna droga usuwania fenyloalaniny jest zablokowana, aminokwas ten występuje we krwi w ilości co najmniej 20 razy większej niż u osób zdrowych.
Fenyloketonuria jest chorobą, w której fenotyp osoby nią dotkniętej warunkowany jest przez zestaw genów oraz czynniki środowiska. Mutacje w genie PAH powodują niepełnosprawność intelektualną tylko w obecności fenyloalaniny w spożywanym przez chorego jedzeniu. Leczenie fenyloketonurii przez eliminację z diety pokarmów zawierających ten aminokwas prowadzi do zmniejszenia jego akumulacji w organizmie człowieka i zapobiega upośledzeniu umysłowemu.
Najważniejsze objawy fenyloketonurii to:
zahamowanie rozwoju psychoruchowego, niepełnosprawność intelektualna (ze względu na zaburzenia procesów komórkowych w mózgu, w tym mielinizacji nerwów oraz syntezy białek);
nadpobudliwość;
małogłowiemałogłowie;
jasna karnacja i jasne włosy (tyrozyna, powstająca z fenyloalaniny, jest substratem do produkcji barwnika melaniny).
Diagnostyka i leczenie
W Polsce częstość fenyloketonurii wynosi 1 : 7000 żywych urodzeń, dlatego u każdego noworodkanoworodka po narodzinach wykonywany jest test przesiewowytest przesiewowy w kierunku tej choroby. Test ten daje szansę na postawienie diagnozy na etapie przedobjawowym i szybkie wdrożenie leczenia, które polega na stosowaniu diety pozbawionej aminokwasu fenyloalaniny. Pozwala to na zapobieganie uszkodzeniu mózgu. Dieta ta musi być kontynuowana przez całe życie.

Alkaptonuria
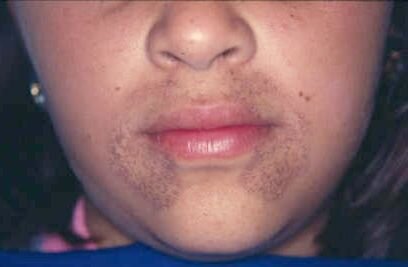
Alkaptonuria to rzadka choroba wywołana nieprawidłową syntezą dioksygenazy 1,2‑homogentyzynowej. Brak lub niedobór tego enzymu prowadzi do wydalania z moczem dużej ilości kwasu homogentyzynowego (HGA), będącego metabolitem pośrednim przemian fenyloalaniny i tyrozyny. Powoduje to ciemnienie moczu w kontakcie z powietrzem. Dodatkowo produkty utleniania HGA odkładane są w tkance łącznej, co przyczynia się do zwyrodnienia stawów i nieprawidłowej pigmentacji skóry.
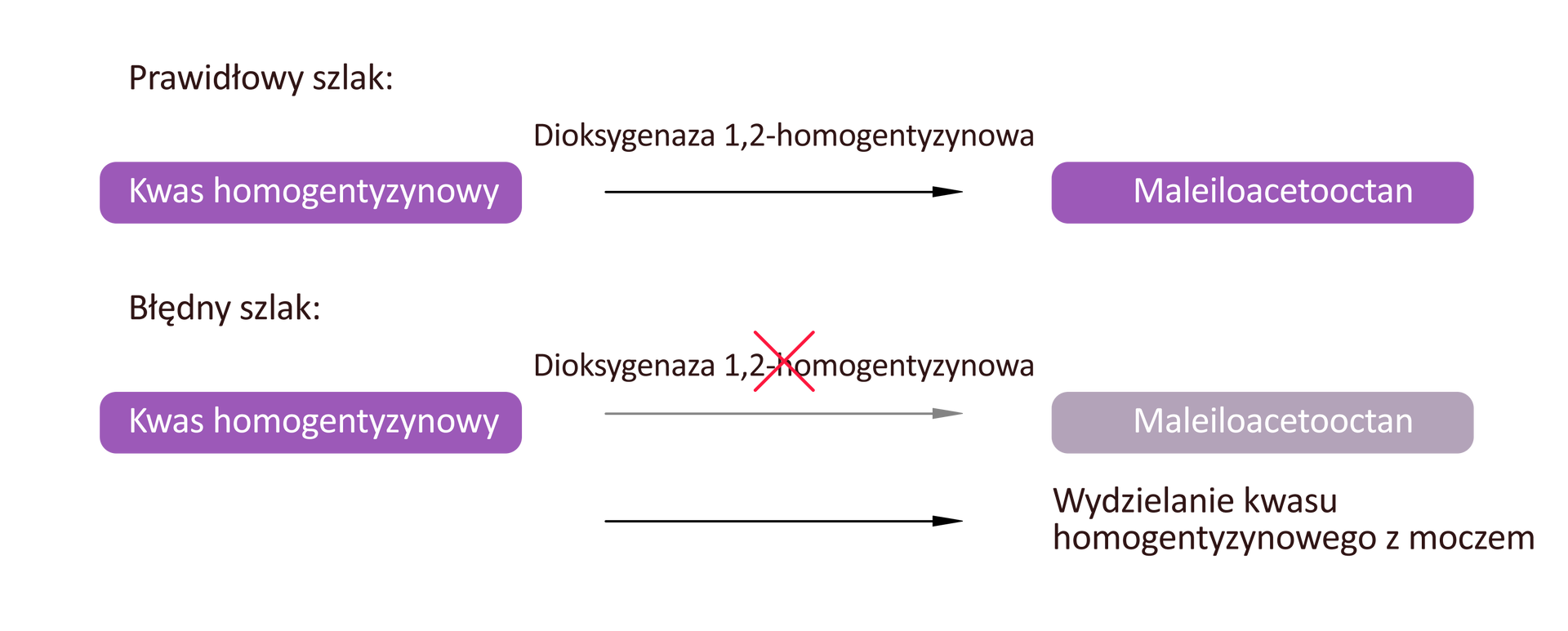
Najważniejsze objawy alkaptonurii to:
zmiany zwyrodnieniowe stawów (tzw. artropatia ochronotyczna);
ochronozaochronoza;
ciemnienie moczu w kontakcie z powietrzem.
Diagnostyka i leczenie
Alkaptonuria diagnozowana jest poprzez dodanie roztworu Fehlinga do moczu. Jeżeli obserwuje się zmianę zabarwienia próbki na szaroczarną, oznacza to, że badana osoba jest chora. Inna metoda diagnostyczna polega na dodaniu do moczu 10‑procentowego roztworu NaOH – u osób chorych prowadzi to do ściemnienia próbki i powstania ciemnego pierścienia.
Leczenie alkaptonurii polega przede wszystkim na utrzymaniu prawidłowej diety. Ważne jest, aby chory ograniczył przyjmowanie białek bogatych w fenyloalaninę i tyrozynę, ponieważ ogranicza to wydalanie kwasu homogentyzynowego w moczu i spowalnia rozwój objawów choroby, takich jak ochronoza. Istotne jest również dobranie odpowiedniego rodzaju sportu dla dziecka, nieobciążającego jego stawów.
Albinizm (bielactwo)
Albinizm wynika z mutacji w genie TYR, który koduje tyrozynazę. Jest ona kluczowym enzymem melanogenezy, który przekształca 3,4‑dihydroksyfenyloalaninę (DOPA) w dopachinon, czyli związek pośredni w biosyntezie melaniny (barwnika skóry). Niedobór lub brak tyrozynazy powoduje blok w szlaku metabolicznym, który w prawidłowych warunkach jest zaangażowany w syntezę barwników melaninowych: eumelaniny i feomelaniny. W konsekwencji u osoby dotkniętej albinizmem występuje bardzo mało tych barwników w skórze, włosach i tęczówkach oczu.

Najważniejsze objawy albinizmu to:

bardzo jasna skóra;
białe włosy, brwi i rzęsy;
niezabarwiona tęczówka oka, przez którą prześwitują naczynia krwionośne;
oczopląs;
zez;
obniżona ostrość wzroku.
Diagnostyka i leczenie
Albinizm diagnozowany jest na podstawie charakterystycznych objawów klinicznych, przede wszystkim wyglądu skóry.
Osoby dotknięte albinizmem powinny odpowiednio chronić się przed słońcem, przede wszystkim poprzez stosowanie filtrów UV oraz noszenie odpowiedniej odzieży ochronnej. Ponadto albinosi powinni raz w roku zgłaszać się na badanie okulistyczne i dermatologiczne w celu wykrycia wczesnych postaci nowotworów.
Inne choroby bloku metabolicznego
Galaktozemia
Galaktozemia to schorzenie spowodowane nietolerancją cukru galaktozy, wynikającą z niedoboru enzymu biorącego udział w jej przekształcaniu (urydylotransferazy galaktozo‑1-fosforanowej). Na skutek jego niedoboru w organizmie gromadzą się galaktoza oraz galaktozo‑1-fosforan – związki te działają toksycznie, m.in. na wątrobę, nerki i soczewkę oka.
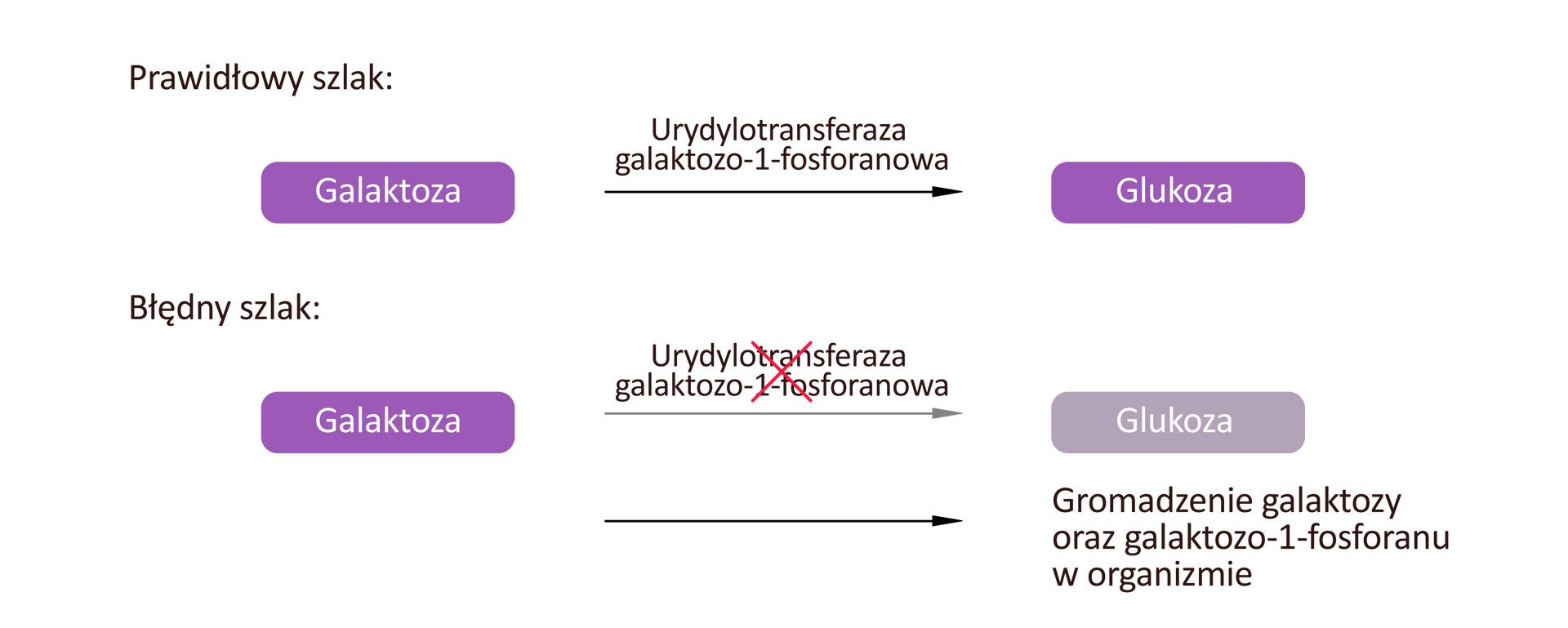
Najważniejsze objawy galaktozemii to:
wymioty, biegunka;
przedłużona żółtaczka noworodkoważółtaczka noworodkowa;
zaćma, czyli zmętnienie soczewki oka;
powiększenie wątroby i śledziony;
objawy uszkodzenia ośrodkowego układu nerwowegoośrodkowego układu nerwowego (zaburzenia chodu, zaburzenia koordynacji ruchowej, upośledzenie umysłowe).
Leczenie jest skuteczne przy wczesnym rozpoznaniu choroby. Polega na stosowaniu diety pozbawionej mleka, które zawiera laktozę będącą dwucukrem złożonym z galaktozy i glukozy, i jego przetworów.
Choroba syropu klonowego
Istotą choroby syropu klonowego jest niska aktywność kompleksu dehydrogenazy alfa‑ketokwasów o rozgałęzionym łańcuchu, który odpowiada za dekarboksylacjędekarboksylację leucyny, izoleucyny i waliny, czyli aminokwasów rozgałęzionych. Nazwa schorzenia wywodzi się od zapachu moczu osoby chorej, przypominającego zapach syropu klonowego.
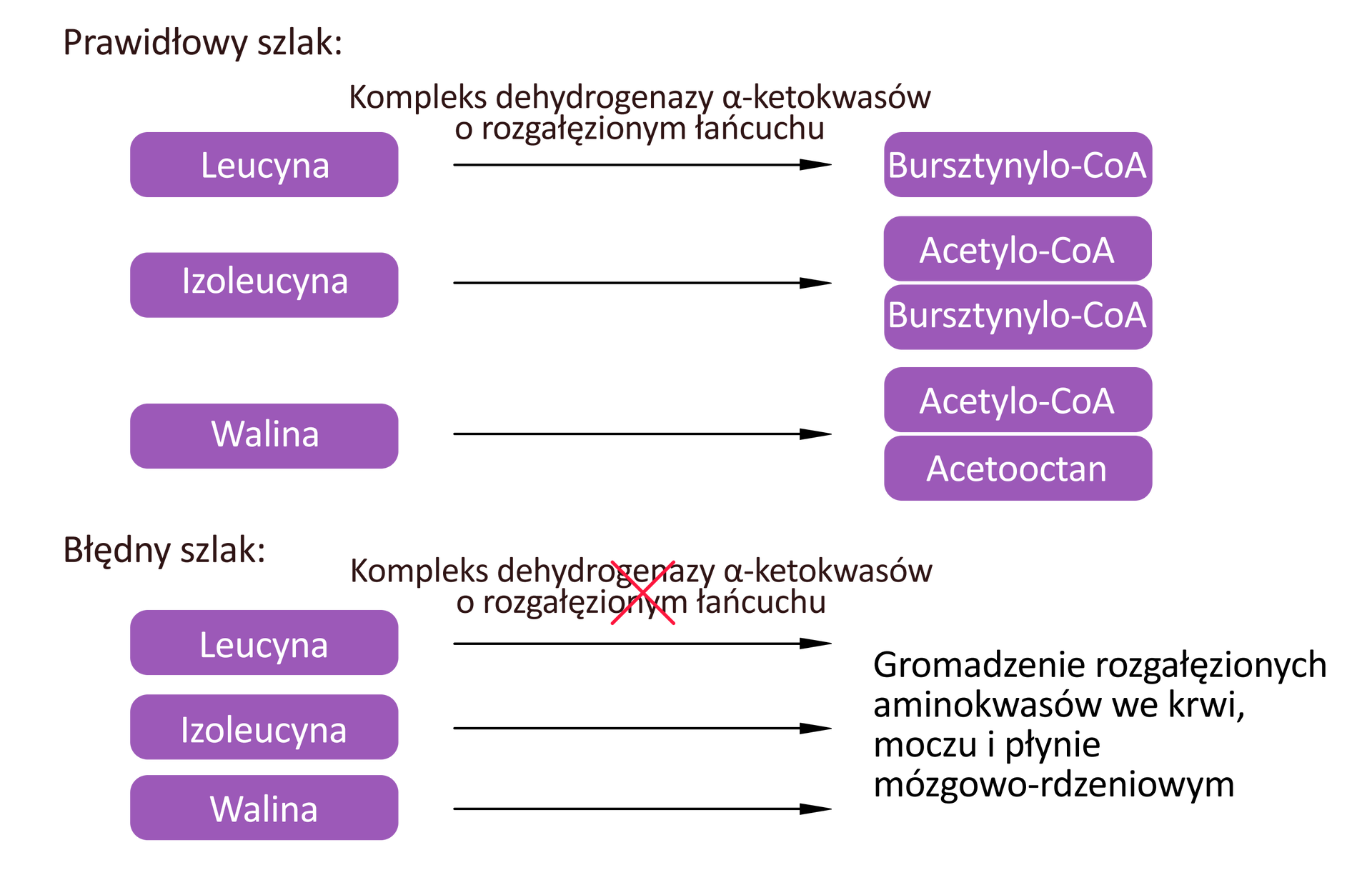
Objawy tej choroby pojawiają się u noworodka kilkanaście dni po urodzeniu.
Najważniejsze objawy choroby syropu klonowego to:
wymioty, nudności;
drżenia;
problemy z karmieniem;
wiotkość;
senność;
uszkodzenie ośrodkowego układu nerwowego prowadzące do śpiączki.
Leczenie polega na stosowaniu diety z wyłączeniem aminokwasów rozgałęzionych (izoleucyny, leucyny, waliny). W stanie ciężkim, wywołanym nagromadzeniem toksycznych produktów, leczenie polega na zastosowaniu dializy.
Fruktozemia
Fruktozemia polega na nietolerancji cukru fruktozy, w wyniku niedoboru enzymu aldolazy fruktozo‑1-fosforanu, który bierze udział w przemianach biochemicznych fruktozy. Objawy kliniczne spowodowane są nagromadzeniem w organizmie fruktozo‑1-fosforanu.
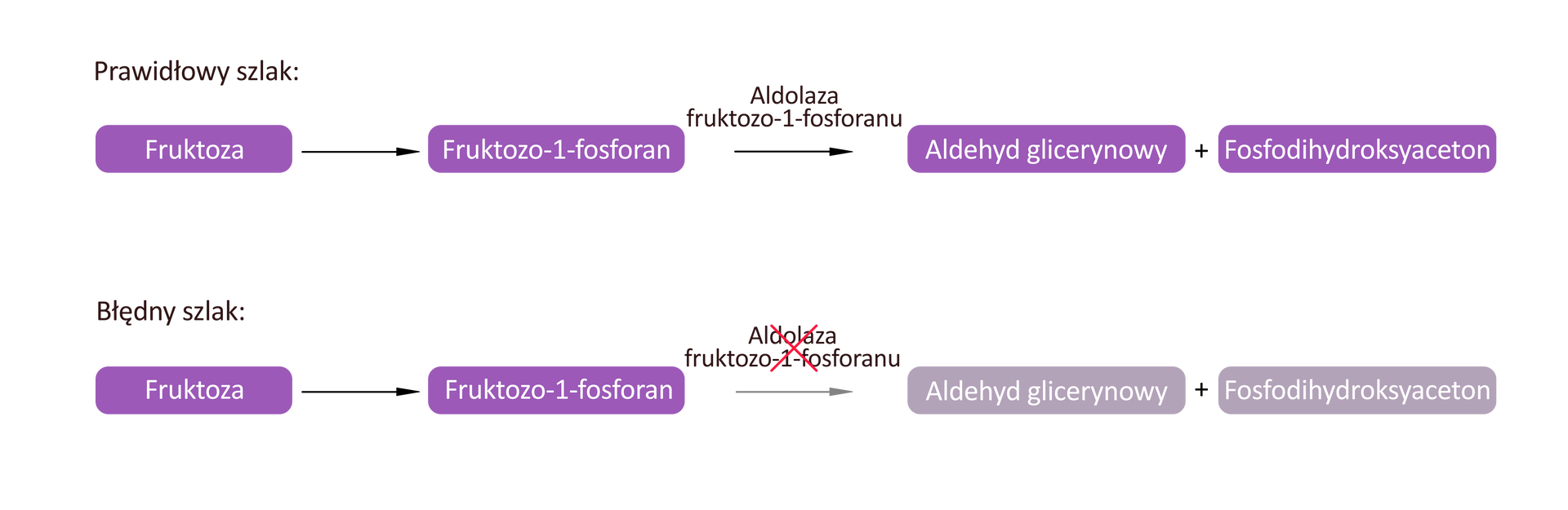
Najważniejsze objawy fruktozemii to:
nudności, wymioty, bóle brzucha (związane z przyjmowaniem pokarmów bogatych we fruktozę);
niechęć do spożywania produktów zawierających fruktozę (owoce, warzywa, słodycze);
zaburzenia wzrastania i przyrostu masy ciała.
Leczenie polega na eliminacji z diety fruktozy, ale również sacharozy, która jest dwucukrem zbudowanym z jednej cząsteczki glukozy i jednej cząsteczki fruktozy.
Słownik
forma genu przejawiająca swój efekt fenotypowy jedynie w układzie homozygotycznym; nie ujawnia się u heterozygot; oznaczany małą literą alfabetu
reakcja chemiczna, w wyniku której dochodzi do usunięcia z danego substratu grupy karboksylowej (–COOH); w reakcji tej produktem jest typowo dwutlenek węgla
aminokwas egzogenny występujący w większości białek; w organizmie zwierzęcym utlenia się do tyrozyny
gen kodujący hydroksylazę fenyloalaninową
enzymy katalizujące reakcje hydroksylacji, czyli dołączenia do danego substratu grupy hydroksylowej (–OH)
zaburzenie neurorozwojowe, polegające na nieprawidłowo małym wymiarze czaszki; może towarzyszyć wielu chorobom o podłożu genetycznym, takim jak zespół Downa, zespół Edwardsa, zespół Pataua
dziecko od urodzenia do 4 tygodnia życia; w tym czasie przystosowuje się ono do życia w zupełnie nowym środowisku
objaw alkaptonurii; odkładanie się polimerów kwasu homogentyzynowego w postaci barwnika w tkance łącznej
część układu nerwowego u kręgowców; tworzony jest przez mózgowie oraz rdzeń kręgowy
badania osób zdrowych mające na celu wykrycie choroby w jej wczesnym, bezobjawowym stadium
aminokwas aromatyczny; składnik wielu białek, występuje również w stanie wolnym; powstaje z egzogennej dla zwierząt fenyloalaniny
stan fizjologiczny, u podstawy którego leżą zaburzenia przemiany bilirubiny; objawia się zażółceniem twarzy, tułowia, kończyn; na samym końcu zabarwieniu ulegają dłonie i stopy